A microscope equipped with 50 mW 405 nm and 100 mW 488, 561 and 642 nm solid-state lasers, an EMCCD 512×512 camera, an alpha Plan Apo 100X/1.46 objective and Band Pass 570-650 / Long Pass 655 emission filters was used for the representative results presented below.
Figure 1A gives an example of molecule density and signal to noise ratio that should be used during raw image acquisition. Good quality images are obtained with a minimum of ~1 µm between neighboring molecules. In good blinking conditions, fluorophores should be present in only one frame. Good blinking requires to adjust the imaging buffer conditions (pH, reductive agent, oxygen scavenging system) and laser powers, which influence the on- and off-rates of the fluorophores, and therefore the duty cycle (the fraction of time a fluorophore spends in the on state)9,11. Figure 1B, on the contrary, gives an example of raw image where the molecule density is too high and single molecule cannot be resolved.
Figure 1C shows a typical histogram of localization precisions for all the molecules detected and analyzed by the manufacturer software from a stack of 20 000 frames. This histogram corresponds to the dSTORM image of the microtubule network shown in Figure 3B. Values of localization precisions provided by the manufacturer software are in good agreement with values estimated following step 8, using the pointing precision of a single molecule localized at different time points22 (Figure 1D).
Figure 2A shows a typical example of dSTORM image of vimentin filaments immunolabeled with Alexa647. The increase in resolution can clearly be observed on comparison with the image acquired with a standard wide-field microscope (Figure 2B-C). The fluorescence intensity profiles represented in Figure 2D show that super-resolution microscopy imaging allows resolving vimentin bundles. The dSTORM resolution is sufficient to count the number of filaments present in the bundles.
Figure 3 shows a typical example of dual-color dSTORM image of the vimentin and tubulin networks immunolabeled with Alexa647 and Alexa555 respectively. The overlay of the two networks shows that the vimentin bundles are often localized along microtubules, providing key structural information on the coupling between the two cytoskeleton subsystems.
Figure 4 illustrates various examples of dSTORM images of microtubules obtained in non-optimal conditions. First, using Alexa488 and a buffer with 143 mM beta-mercaptoethanol (another reductive agent used instead of MEA9,10,11) and an oxygen scavenging system (composed of 0.5 mg/mL glucose oxidase and 40 µg/mL catalase), the fluorophores were blinking but were not bright enough to be precisely localized (too low photon number) (Figure 4A). Second, using Alexa555 with 143 mM beta-mercaptoethanol, 50 mM MEA and an oxygen scavenging system, the fluorophores could not be pumped to their dark state and therefore not be well spatially separated (too high duty cycle, i.e. the fraction of time in the "on" state is too long) (Figure 4B). Finally, using Alexa568, with 143 mM beta-mercaptoethanol, 50 mM MEA and an oxygen scavenging system, the fluorophores were not bright and were blinking slowly with very long "on" and "off" states (low photon number and too high duty cycle) (Figure 4C). In all these non-optimal conditions, the microtubule network was poorly rendered in the super-resolution images. In conclusion, optimal conditions require fluorophores with high photon number and low duty cycle9,10 in the corresponding imaging buffer. Note that optimizing labeling density and imaging buffer conditions to obtain good blinking conditions is a standard procedure for dual-color dSTORM and is not specific to IF-microtubule imaging.
According to the Nyquist-Shannon theorem, it is necessary to have fluorophores at least every 10 nm to have a resolution of 20 nm. Extended to 2D structures, Dempsey et al estimated that a labeling of ~104 fluorophores per µm2 is necessary. As the IF network is denser and its filaments are thinner (10 nm vs 25 nm diameter without the primary and secondary antibodies) compare to the microtubule network, we observed that twice more localizations are necessary to describe the IF network. We obtained good images with 5 000 localizations/µm2 for IF and 2500 localizations/µm² for microtubules, obtained with 40 000 frames and 20 000 frames respectively. The image with the highest resolution was obtained with a localization precision of σSMLM ~ 8-12 nm estimated following step 8 of the protocol (pointing precision of a single molecule localized at different time points22). This estimation of localization precision was in good agreement with the values provided by the manufacturer software shown in a histogram of localization precisions extracted from all the detected molecules in the raw data (example shown in Figure 1C). Such localization precision could provide an image with a resolution of ~30 nm (2.35*precision) if the labeling density is high enough. We routinely observed vimentin filament with a Full Width at Half Maximum (FWMH) of ~40 nm and microtubules with a width FWMH of ~55 nm. Since the image resolution depends on both the localization precision and the localization density, we provide experimental conditions which allow the best results taking into account both criteria.
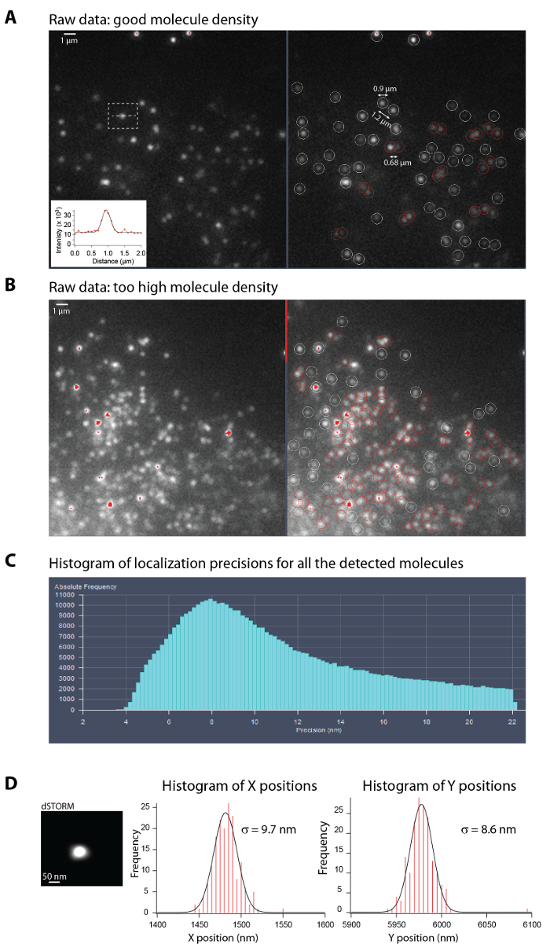
Figure 1: Optimal density of single molecules during acquisition and localization precision estimation.
(A-B) Left: Representative raw images from a single frame with an optimal density of single molecules in the bright state (A) and with a too high density (B) during STORM acquisition (step 6.12.) of fixed U373 cells stained with anti-vimentin and anti-mouse Alexa647. Right: Raw images where the single molecules with fluorescence level above the Peak intensity to noise threshold (set to 6) are surrounded by circles (white or red). The circle diameter is set by the Peak mask size (set to 9 pixel) and the color determines whether the molecules overlap (red) or not (white). Note that high density of single molecules (in (B)) lead to a high amount of overlapping molecules which are not taken into account for the STORM image reconstruction. A Inset: Graph showing a typical fluorescence intensity profile of a single molecule in red and its Gaussian fit in black. In this example, the localization precision is σx = 6.7 nm. Scale bar, 1 µm. (C) Typical histogram of localization precisions for all the molecules detected and analyzed by the manufacturer software. It can be visualized and is updated regularly during the acquisition of the raw data thanks to the online processing. This histogram corresponds to the dSTORM image of the microtubule network shown in Figure 3B. (D) Left: STORM image of a single molecule presents on the glass surface after reconstruction. Right: Histograms of the X and Y positions obtained after Gaussian fitting. Gaussian fitting the X and Y distributions give an estimate of the localization precision. Please click here to view a larger version of this figure.
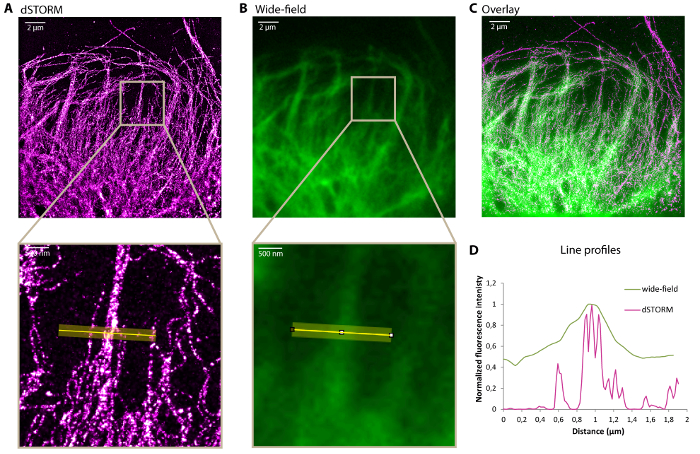
Figure 2: Representative dSTORM image of a vimentin network. dSTORM image (A), corresponding standard wide-field image (B) and overlay (C) showing the leading edge of a U373 glial cell fixed as described in the protocol and stained for vimentin with Alexa647. Bottom: zoom of the highlighted region. (D) Transverse intensity profiles along the yellow line on the zoomed images in A and B. Scale bar, 2 µm and 500 nm in the zoom. Please click here to view a larger version of this figure.
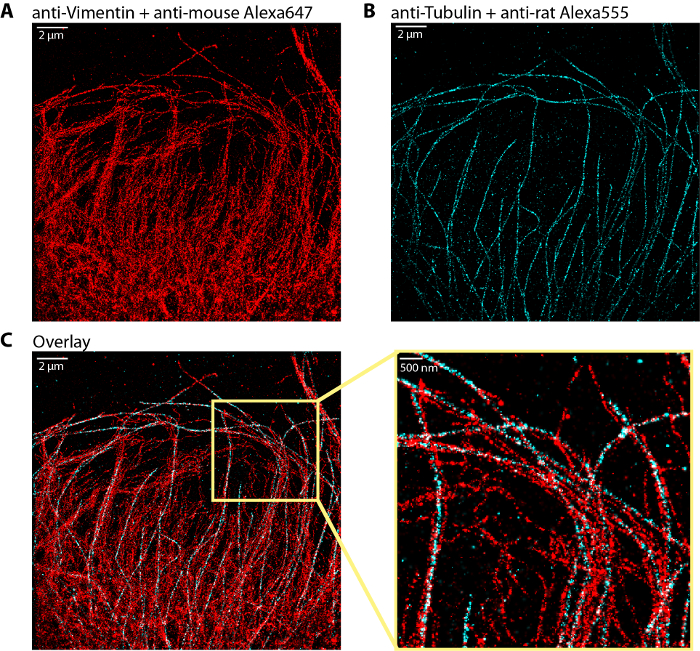
Figure 3: Representative dual color dSTORM image of vimentin and microtubule networks.(A) Vimentin network stained with Alexa647 (same as in Figure 2A). (B) Microtubule network labeled with Alexa555. (C) Overlay of the vimentin (red) and microtubule (cyan) networks with a zoom region on the right. The dSTORM image of vimentin was obtained with ~1.5 million of localizations out of 40 000 frames. The dSTORM image of microtubules was obtained with ~850 000 localizations out of 20 000 frames. Scale bar, 2 µm and 500 nm in the zoom. Please click here to view a larger version of this figure.
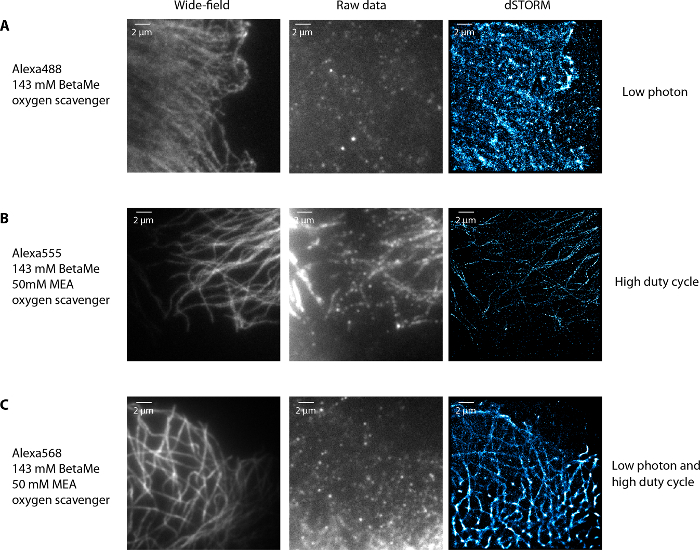
Figure 4: Examples of non-optimal imaging conditions of microtubules. When the fluorophores blink but are not bright enough with an Alexa488 labeling of tubulin in U373 glial cells and 143 mM BetaMercaptoethanol and oxygen scavenger in the imaging buffer (A), when the fluorophores are bright enough but cannot be properly depleted in the dark state (at the maximum laser power available) and blink slowly with Alexa555 labeled tubulin in 143 mM BetaMercaptoethanol, 50 mM MEA and oxygen scavenger buffer (B) and when fluorophores blink slowly and are not bright using Alexa568 labeled tubulin in a storm buffer that contains 143 mM BetaMercaptoethanol, 50 mM MEA and oxygen scavenger (C). In all these cases, the dSTORM images have degraded resolution. Scale bar, 2 µm. Please click here to view a larger version of this figure.
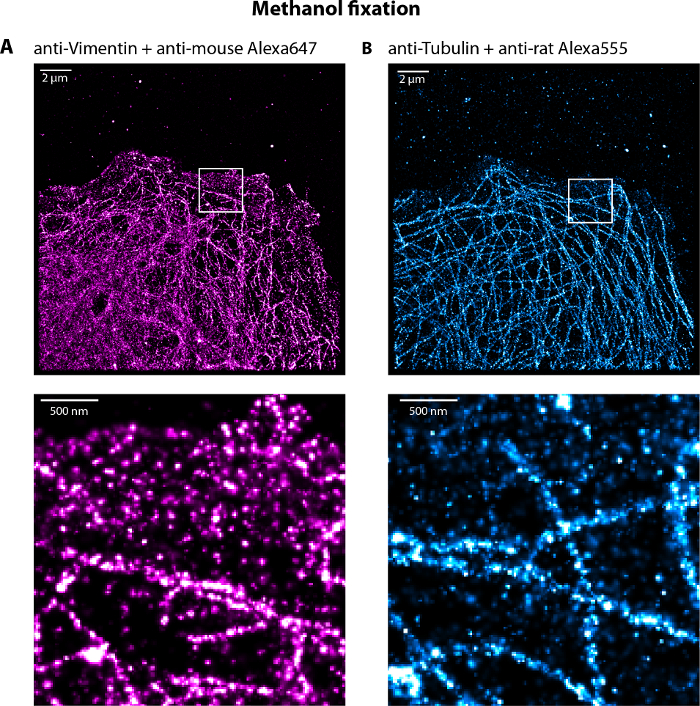
Figure 5: Representative dual-color dSTORM image of vimentin and microtubule networks of a U373 cell fixed with cold methanol for 5 min. (A) Vimentin and (B) microtubules networks labeled with Alexa647 and Alexa555 respectively with highlighted zooms. Note the presence of single molecules at the front of the cells localized in the cytoplasm decreasing the global resolution of the filament networks. Scale bar, 2 µm and 500 nm in the zoom. Please click here to view a larger version of this figure.
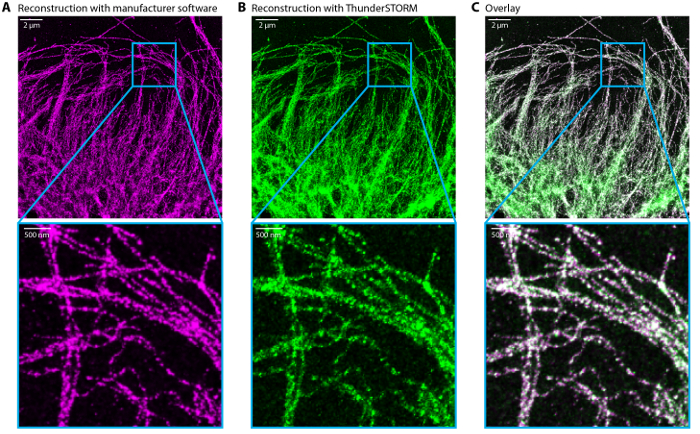
Figure 6: Comparison of dSTORM reconstructed images
Reconstructed dSTORM image by the manufacturer software (A) and ThunderSTORM (FiJi plugin) (B). (C) Overlay of (A) in magenta and (B) in green. Scale bar, 2 µm and 500 nm in the zoom. Please click here to view a larger version of this figure.
