This method generates biofilm thin-sections wherein distinct morphological features and zones of gene expression can be imaged by DIC, fluorescence microscopy, and TEM. While DIC imaging using a 40X oil immersion objective can be sufficient to show some morphological features (Figure 2E), we have found that fluorescence microscopy of strains engineered to constitutively express fluorescent protein provides enhanced visualization of cell distribution within the sample (Figure 2D). Images of individual sections can be stitched together to generate a cross-section of the entire colony (Figure 2B) and to provide context for the localization of structural features within the overall morphology at the macroscopic level (compare Figure 2A, B, and C). Morphological features and fluorescent signals can be measured using imaging software14. Structures within the biofilm or transitions between zones of gene expression can then be correlated with distributions of metabolites or chemical gradients11 .
This protocol can be amended and adapted in several ways. Use of a strain engineered to express fluorescent protein under the control of a specific promoter enables visualization of the distribution of gene expression (Figure 3A). Colonies can be grown on medium containing dyes, or dyes can be added post-sectioning, to stain specific polysaccharides (Figures 3B and C). Finally, samples prepared in a modified version of this protocol can be examined by TEM (Figure 3D) to allow for visualization at higher resolution than that enabled by DIC or fluorescence microscopy.
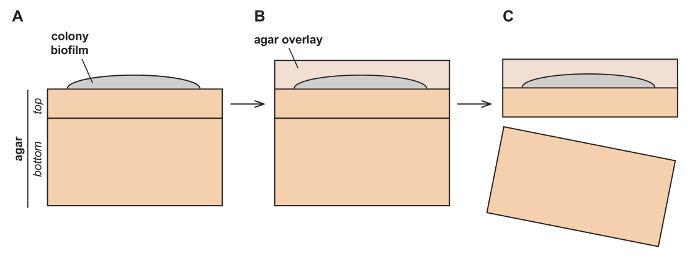
Figure 1: Schematic depicting setup for colony growth prior to fixation.
(A) Colonies are grown on top of two layers of agar-solidified growth medium (top and bottom) and (B) are then overlaid with an additional layer (overlay), which preserves colony morphology. (C) The colony sandwiched between the overlay and top layer is then separated from the bottom layer for further processing. Please click here to view a larger version of this figure.
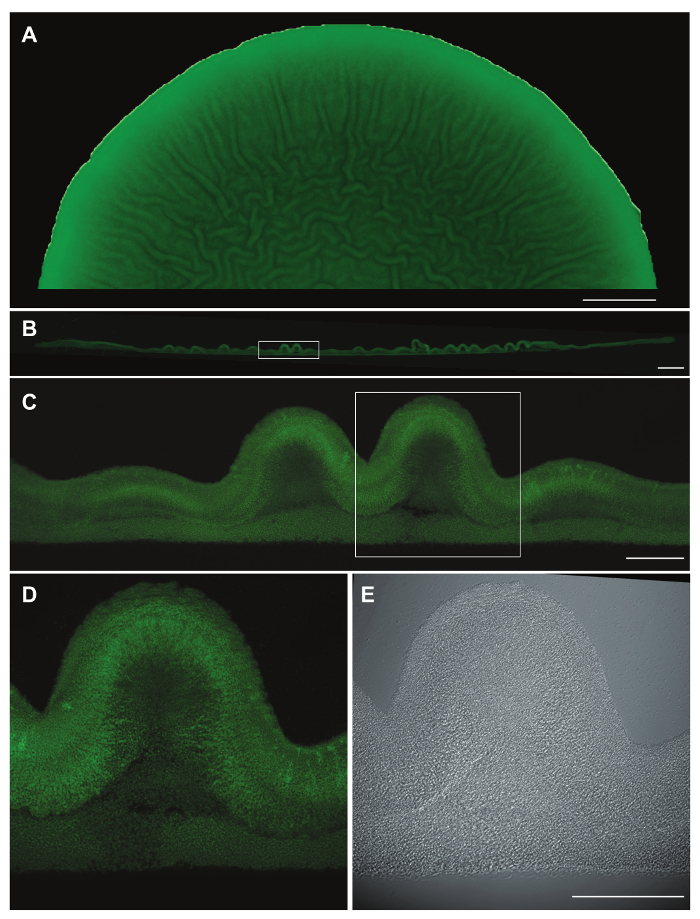
Figure 2: Micromorphological features and YFP fluorescence in cross-sections of paraffin-embedded colonies.
Representative data for a Pseudomonas aeruginosa PA14 Δphz colony15 constitutively expressing YFP and grown for 3 days on 1% tryptone, 1% agar containing Congo red and Coomassie blue. (A) Top-view, fluorescence image showing half of a colony prior to fixation. (B) Cross section of the entire colony after paraffin-embedding. The boxed area was imaged with a 10X objective lens (C). The boxed area in (C) was then imaged with a 40X oil immersion objective lens using fluorescence (D) and DIC (E). Scale bars represent 2 mm (A), 500 µm (B), and 100 µm (C-E). Please click here to view a larger version of this figure.
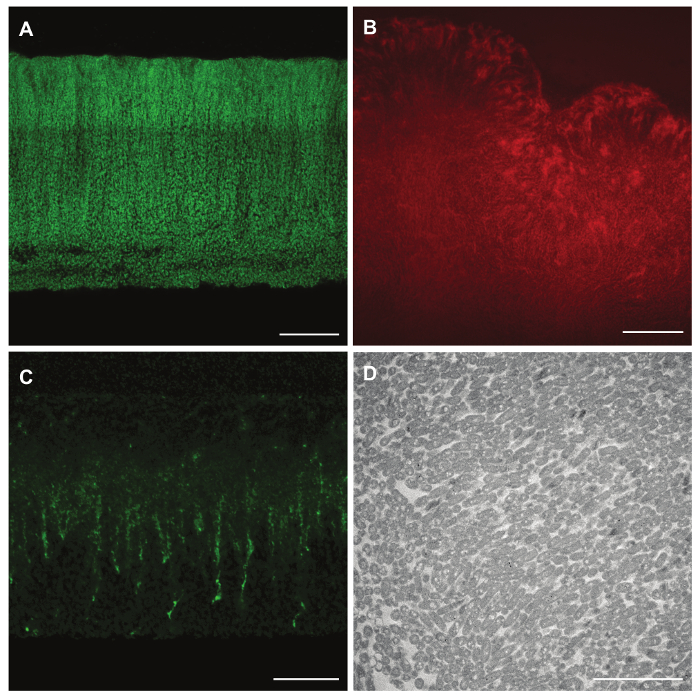
Figure 3: Reporter expression, pre- and post-staining of biofilm features, and TEM imaging of paraffin-embedded colony biofilm samples.
(A) mexGPr-GFP reporter fluorescence in a P. aeruginosa PA14 colony grown for 3 days on 1% tryptone, 1% agar. (B) Fluorescence of bound Congo red in a P. synxantha colony grown for 5 days on a medium containing the dye. (C) Fluorescence of the Wisteria floribunda lectin stain for Pel polysaccharide in a P. aeruginosa PA14 Δphz colony (grown for 2 days on 1% tryptone, 1% agar containing Congo red and Coomassie blue), applied after sectioning. (D) TEM image of a P. aeruginosa PA14 Δphz colony (grown for 3 days on 1% tryptone, 1% agar containing Congo red and Coomassie blue), and processed for sectioning via paraffin embedding*. Scale bar represents 40 µm (A-B), 20 µm (C), and 5 µm (D). Please click here to view a larger version of this figure.
*This sample was processed for sectioning via paraffin-embedding through Step 4 of the above protocol, omitting Steps 4.1-4.3, and then processed separately for TEM analysis.
