The goal of this protocol is to perform gene knockout (KO) in mouse and human HSPCs using Cas9-sgRNA RNPs. The workflow is easy and fast and allows for completion of experiments in less than a week from experimental design to assessment of KO efficiency.
Compared to plasmid- or virus-based approaches, the success of our protocol builds upon the combination of sgRNA RNPs and electroporation. This cloning-free strategy significantly shortens the time needed to obtain the components to transfect. Moreover, electroporation allows for transfection of up to 95% of cells, maximizing the delivery of the RNPs. sgRNAs can be synthetized in the lab through in vitro transcription (IVT), while purified Cas9 protein can be purchased from several companies (see protocol). sgRNA DNA templates for IVT are obtained performing a short PCR using the sgRNA Fwd primer (Figure 1A, B) and the universal scaffold Rev primer (Figure 1B – see Protocol). The PCR product is then used as a template for the IVT (Figure 1C). Cas9-sgRNA RNP complexes are generated incubating for 15 – 30 minutes the sgRNAs and the Cas9 (Figure 1C). Finally, sgRNA RNPs are electroporated into cells. Edited HSPCs can then be used to perform both in vitro and in vivo experiments.
Gene disruption in mouse cells
To demonstrate the efficacy of the Cas9-sgRNA RNP electroporation strategy, c-kit+ HSPCs isolated from mice ubiquitously expressing GFP or tdTomato have been electroporated with sgRNA against GFP or tdTomato. Among the three sgRNA designed against GFP, GFP sg1 performed most efficiently, exhibiting approximately 70 – 75% disruption of GFP expression as determined by flow cytometry (Figure 2A, B). To quantify the gene disruption frequency at the genomic level, genomic DNA from the electroporated cells was isolated and T7 endonuclease I-based (T7EI) assay was performed. As shown in Figure 2C, T7EI assay detected up to 60% indel formation. Note that T7EI assays tend to underestimate the frequencies of indels. We also generated sgRNA against tdTomato and electroporated HSPCs expressing tdTomato from the Rosa26 locus. As shown in Figure 2D, tdTomato sg1 efficiently ablated the expression of tdTomato.
Gene disruption in human cells
Here, efficient disruption obtained with a single sgRNA approach targeting CD45, a cell surface marker expressed on hematopoietic cells, is shown. Three sgRNAs targeting different exons of CD45 were designed (CD45 sg1, sg2, and sg3). The efficiency of the sgRNAs was tested in the HL60 AML cell line (Figure 3A). Protein KO was assessed by flow cytometry 5 days after electroporation. CD45 sg1 was the most efficient (98% KO efficiency – Figure 3A) and was used for further experiments. Figure 3B shows CD45 KO in primary human CD34+ progenitor cells using CD45 sg1, assessed by flow cytometry 5 days after electroporation. While CD34 expression was unaffected, CD45 expression was lost in about 80% of the cells. 200.000 edited primary CD34+ cells were retro-orbitally transplanted into NSG mice 6 hours after electroporation. Bone marrow and spleen cells were harvested 3 months after transplant. Edited cells (HLA-ABC positive, CD45 negative, highlighted in red) are detectable only in sgRNA treated samples and not in Cas9 only controls (Figure 3C).
If desired, two sgRNAs targeting nearby sequences can be used to generate deletions. This approach allows for a fast estimate of the editing efficiency with a PCR and often produces higher disruption efficiency. Figure 3D shows the efficient generation of a 58 bp deletion in exon 10 of DNMT3A. 200.000 edited CD34+ cells were transplanted into NSG mice 6 hours after electroporation. The 58 bp deletion was clearly detected in the peripheral blood of engrafted NSG mice 5 months after injections (Figure 3E) confirming the editing of CD34+ cells with long-term engraftment capabilities.
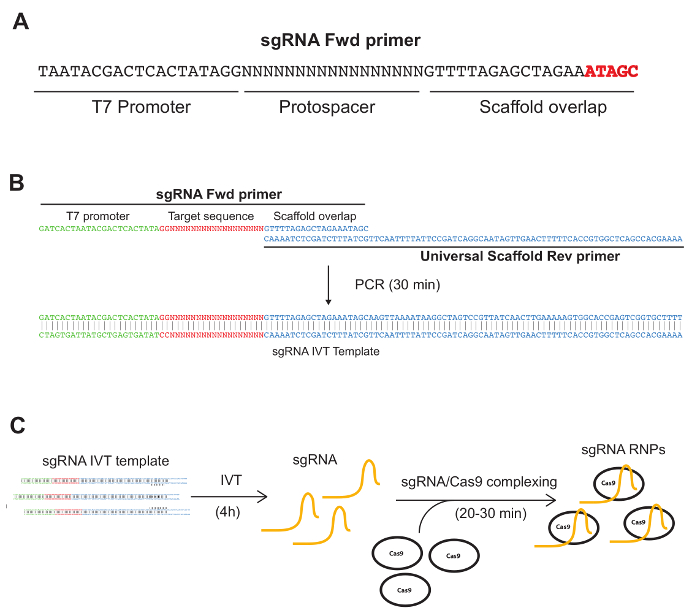
Figure 1: The fast and easy approach to generate sgRNA RNPs. A) Representation of the sgRNA Fwd primer. The sgRNA Fwd primer is a DNA oligonucleotide containing the T7 promoter, the protospacer sequence and an overlap sequence with the sgRNA scaffold. At the 3' end, highlighted in red, is the "ATAGC" sequence that needs to be added to the sgRNA Fwd primer sequence obtained on CRISPRscan. B) Schematic representation of the overlap PCR performed to obtain the IVT template. The overlap between the sgRNA Fwd primer and the Universal Scaffold Rev primer allows for the extension and amplification by PCR. C) Cartoon describing the IVT reaction and the sgRNA RNP pre-complexing. The PCR product is purified and used as a template for the IVT. IVT generates a high amount of sgRNAs that can be used in multiple replicates (see protocol). sgRNA RNPs are generated incubating the sgRNA purified from IVT and the previously purchased Cas9 protein. Please click here to view a larger version of this figure.
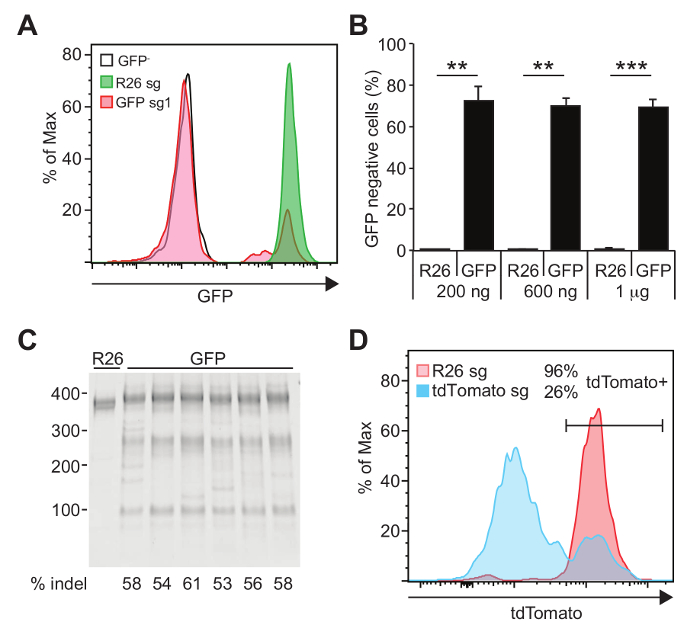
Figure 2: Efficient gene disruption in mouse hematopoietic progenitor cells. A) An sgRNA targeting GFP (GFP sg1) together with Cas9 protein was electroporated into murine c-kit+ HSPCs expressing GFP, and analyzed by flow cytometry 24 hours after electroporation. B) Different amount of GFP sg1 was complexed with 1 µg of Cas9 protein and electroporated. As little as 200 ng of GFP sg1 efficiently ablated GFP expression. C) T7EI assay performed on genomic DNA isolated from cells used in A-B. PCR amplicon spanning the Cas9-sgRNA cleavage site was diluted 1:4 in 1x Buffer 2 and hybridized slowly in a thermal cycler. Hybridized fragments were then digested with 1.25 U of T7 endonuclease I for 10 minutes at 37 °C. Digested fragments were separated by polyacrylamide gel electrophoresis. Band intensities were analyzed using ImageJ software by plotting band intensities of each lane. % cleavage was calculated by the ratio of the intensities of the cleaved bands to uncleaved bands. D) c-kit+ HSPCs isolated from mice expressing tdTomato was electroporated with Cas9 protein complexed with sgRNA against tdTomato. Flow cytometry performed 24 hours after electroporation revealed that approximately 74% of cells deleted tdTomato. R26 = Rosa26. * p <0.05, ** p <0.01, *** p < 0.001. This figure has been adapted from Gundry et al.6 Please click here to view a larger version of this figure.
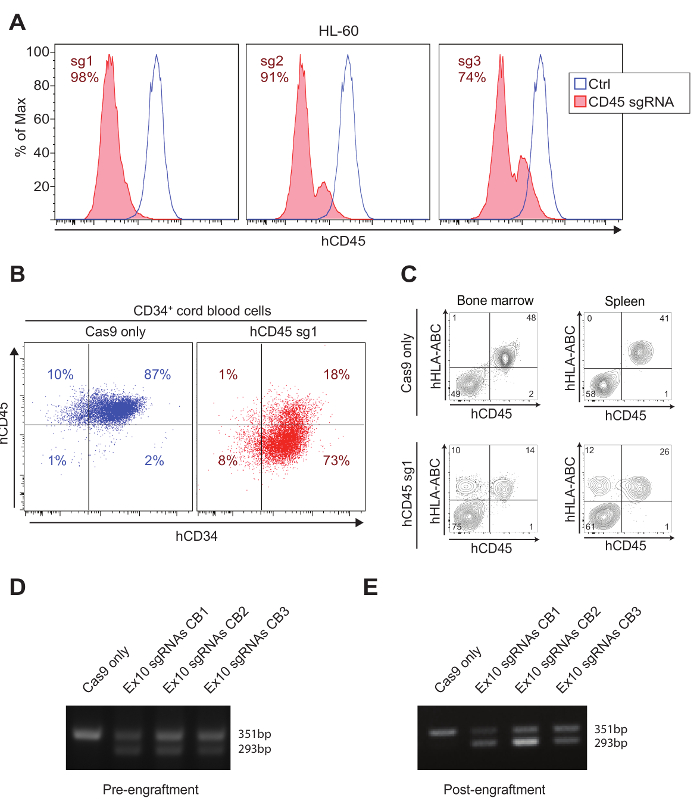
Figure 3: Efficient gene disruption in human hematopoietic cells. A) Three sgRNAs targeting CD45 (CD45 sg1, sg2, and sg3) were designed and tested in the HL60 AML cell line (see protocol for electroporation conditions). Flow cytometry was performed 5 days after electroporation. CD45 sg1 was selected as the most efficient among the three sgRNAs (KO efficiency 98%). B) CD45 KO in primary human cord blood CD34+ HSPCs using CD45 sg1. CD45 loss can be detected in roughly 80% of the cells. Note that CD34 expression is unchanged after editing. C) Edited human HSPCs can be efficiently engrafted into NSG mice. Human nucleated cells display the normal HLA+/CD45+ phenotype in Cas9-only engrafted mice, whereas in mice engrafted with CD45-sg1 treated CD34+ cells, both HLA+/CD45+ and HLA+/CD45- populations are observed indicating engraftment of human CD45 KO cells. D) 2% agarose gel showing a 58 bp deletion generated by 2 sgRNAs (dual guide approach) in exon 10 of DNMT3A in human HSPCs from 3 different cord blood (CB1, CB2, and CB3). PCR was performed 3 days after the electroporation. E) 2% agarose gel demonstrating the persistence of the 58 bp deletion 5 months after transplantation into NSG mice. This figure has been adapted from Gundry et al.6 Please click here to view a larger version of this figure.
| Oligonucleotide | Sequence | Note | ||
| Universal Rev scaffold primer | agcaccgactcggtgccactttttcaagttgataacggactagccttattttaacttgctatttctagctctaaaac | Universal reverse primer for overlap PCR | ||
| hCD45 sgRNA 1 | taatacgactcactataGGTGCTGGTGTTGGGCGCACgttttagagctagaaatagc | sgRNA Fwd primer sequence for overlap PCR | ||
| hCD45 sgRNA 2 | taatacgactcactataGGGAGCAAGTGAGGATCCTCgttttagagctagaaatagc | sgRNA Fwd primer sequence for overlap PCR | ||
| hCD45 sgRNA 3 | taatacgactcactataGGGATGCTTGTTCCCTTCAGgttttagagctagaaatagc | sgRNA Fwd primer sequence for overlap PCR | ||
| hDNMT3A Ex10 sg1 | taatacgactcactataGGACACTGCCAAGGCCGTGGgttttagagctagaaatagc | sgRNA Fwd primer sequence for overlap PCR | ||
| hDNMT3A Ex10 sg2 | taatacgactcactataGGGTGGCCAGCAGCCGCGCGgttttagagctagaaatagc | sgRNA Fwd primer sequence for overlap PCR | ||
| Rosa26 sgRNA | taatacgactcactataGGACACTGCCAAGGCCGTGGgttttagagctagaaatagc | sgRNA Fwd primer sequence for overlap PCR | ||
| GFP sg1 | taatacgactcactataGGGCGAGGAGCTGTTCACCGgttttagagctagaaatagc | sgRNA Fwd primer sequence for overlap PCR | ||
| tdTomato sg1 | taatacgactcactataGGGGTACAGGCGCTCGGTGGgtttaagagctagaaatagc | sgRNA Fwd primer sequence for overlap PCR | ||
Table 1: Complete sequences of sgRNA Fwd primers used in the experiments and Universal Rev primer.
