Generation of Native, Untagged Huntingtin Exon1 Monomer and Fibrils Using a SUMO Fusion Strategy
Summary
Here, we present a robust and optimized protocol for the production of milligram quantities of native, tag-free monomers and fibrils of the exon1 of the Huntingtin protein (Httex1) based on the transient fusion of small ubiquitin related modifier (SUMO).
Abstract
Huntington's Disease (HD) is an inherited fatal neurodegenerative disease caused by a CAG expansion (≥36) in the first exon of the HD gene, resulting in the expression of the Huntingtin protein (Htt) or N-terminal fragments thereof with an expanded polyglutamine (polyQ) stretch.
The exon1 of the Huntingtin protein (Httex1) is the smallest Htt fragment that recapitulates many of the features of HD in cellular and animal models and is one of the most widely studied fragments of Htt. The small size of Httex1 makes it experimentally more amenable to biophysical characterization using standard and high-resolution techniques in comparison to longer fragments or full-length Htt. However, the high aggregation propensity of mutant Httex1 (mHttex1) with increased polyQ content (≥42) has made it difficult to develop efficient expression and purification systems to produce these proteins in sufficient quantities and make them accessible to scientists from different disciplines without the use of fusion proteins or other strategies that alter the native sequence of the protein. We present here a robust and optimized method for the production of milligram quantities of native, tag-free Httex1 based on the transient fusion of small ubiquitin related modifier (SUMO). The simplicity and efficiency of the strategy will eliminate the need to use non-native sequences of Httex1, thus making this protein more accessible to researchers and improving the reproducibility of experiments across different laboratories. We believe that these advances will also facilitate future studies aimed at elucidating the structure-function relationship of Htt as well as developing novel diagnostic tools and therapies to treat or slow the progression of HD.
Introduction
Htt is a 348 kDa protein and has been implicated in several physiological functions1. When Htt contains an expanded polyQ region of more than 36 residues in its N-terminus, it causes HD2,3. HD pathology is characterized by cellular inclusions in the striatum and cortex, which leads to neuronal death and atrophy of the affected tissues4,5. Several N-terminal Htt fragments that contain the polyQ repeat tract have been detected in post-mortem brains from HD patients and are thought to be generated by proteolytic processing of the huntingtin protein6. Recent studies suggest that Httex1 could also be formed due to aberrant mRNA splicing. Httex1 contains the pathological polyQ mutation and its overexpression in animals can recapitulate many of the key features of HD7, thus highlighting a possible central role of this fragment in HD pathology and disease progression6,8,9.
Due to the high aggregation propensity of mutant Httex1 (mHttex1) with expanded polyQ tract, the majority of existing expression systems are based on the transient fusion of Httex1 to proteins (such as glutathione-S-transferase (GST), thioredoxin (TRX) or maltose-binding-protein (MBP) and/or peptides (poly-histidine) that differentially improve its expression, stability, purification and/or solubility10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28. The fusion partner is linked to Httex1 with a short sequence containing a cleavage site for proteases such as trypsin, tabacco etch virus (TEV) protease or PreScission to allow for the cleavage and release of Httex1 prior to the initiation of aggregation or purification. Shortcomings of these methods include the possibility of leaving additional residues due to non-traceless cleavage and the creation of truncated fragments due to miscleavage within the sequence of Httex1, in addition to heterogeneity due to incomplete cleavage (see Vieweg et al. for more in-depth discussion on the advantages and limitations of this approach)10. To address these limitations, we recently developed an expression strategy enabling the generation of tag-free native Httex1 for the first time by utilizing a transient N-terminal fusion of the Synechocystis sp. (Ssp) DnaB intein to Httex110. While the intein cleavage is traceless and specific and yields mg quantity of proteins, it still suffers two drawbacks that could reduce the yield: namely, premature cleavage of the intein which can occur during the expression, and the fact that cleavage occurs over several hours, which could lead to loss of protein due to aggregation, especially for Httex1 with expanded polyQ repeats.
To address these limitations and to refine our strategy for the production of native, tag-free Httex1, we developed a new expression system based on the transient fusion of SUMO, more exactly the yeast homolog Smt3 to Httex1. The application of the SUMO system for the production of recombinant proteins was first published in 200429, where an increased rate of expression and solubility of SUMO fusion protein was demonstrated. The SUMO tag can be cleaved by the ubiquitin like protein-specific protease 1 (ULP1), which does not require a recognition site, but recognizes the tertiary structure of SUMO and practically eliminates the possibility of miscleavage30. Furthermore, the ULP1-mediated cleavage is fast and traceless and does not leave additional residues behind. The premature cleavage of the fusion tag, as observed with the autocatalytic intein10, is completely avoided by the requirement of an external protease. While the SUMO strategy is nowadays widely used for recombinant protein production31,32,33, we demonstrate in this paper that it is especially useful for the generation of an intrinsically disordered, aggregation-prone, amyloidogenic protein such as Httex1. We believe that the simplicity, efficiency and robustness of our SUMO-fusion-based method will make native, tag-free Httex1 more accessible to researchers from different disciplines and eliminate the need to use non-native sequences of Httex1 in vitro. This is an important advance that will facilitate future studies to elucidate the structure-function relationship of Httex1.
The protocol describes the purification of Httex1 from 12 L of bacterial culture, but the protocol could be easily adapted for smaller or larger scale productions. The protocol describes the production of wild type Httex1 (wtHttex1) with a polyQ repeat length below (23Q) and mutant Httex1 (mHttex1) with a polyQ repeat length above (43Q) the pathogenic threshold (36Q).
Protocol
1. Expression of Recombinant Httex1 23Q and 43Q
- Prepare the required buffers and solutions. Prepare 1000x ampicillin (AMP, 100 mg/mL) stock solution, filter (0.2 µm), aliquot and store at -20 °C. Prepare lysogeny broth (LB) medium (25 g LB Miller per 1 L H2O), autoclave. Prepare 1 M Isopropyl ß-D-1thiogalactopyranoside (IPTG) stock solution, filter (0.2 µm), aliquot and store at -20 °C.
- Transform chemical competent E. coli B ER 2566 with a pTWIN1 vector, containing human Httex1 fused to an N-terminal His6-SUMO tag with the heat shock method34.
NOTE: The E. coli BL21 DE3 strain has also been used. However, in this case an increased amount of truncations was observed. - Inoculate 200 mL of LB-medium with 1x AMP in a 1 L conical flask by adding a single colony from the agar plate with a sterile pipette tip. Incubate the culture at 30 °C and 180 rpm for 20 h (overnight) in a bacterial incubator.
- Take a 1 mL sample of the culture with a sterile pipette. Measure the optical density at 600 nm (OD600) of the sample with a disposable plastic cuvette and a photometer (respect the measurement range between 0.1 and 1, dilute with LB-medium if necessary). Calculate the amount of preculture that will result in a starting OD600 of 0.05 in a 3 L culture (with a preculture of OD600 = 3 that would mean 50 mL).
- Inoculate four cultures (each 3 L of LB-medium with 1x AMP in a 5 L flask), by adding the calculated amount of preculture with a sterile pipette. Incubate the cultures at 37 °C and 180 rpm in a bacterial incubator.
- Every 30 min, take a 1 mL sample of the culture with a sterile pipette. Measure the OD600 of the sample with a disposable plastic cuvette and a photometer. When OD600 has reached 0.1 (typically after 1-2 h), set the temperature of the bacterial incubator to 14 °C and continue the incubation while cooling. Every 30 min, take a 1 mL sample of the culture with a sterile pipette. Measure the OD600 of the sample with a disposable plastic cuvette and a photometer.
NOTE: The time to cool the cultures may vary with the incubator used, so the time to begin cooling might have to be adapted depending on the type of incubator used. However, changing the temperature gradient should only have a small impact on the yield as the SUMO fusion protein appears to be quite stable. - When OD600 has reached 0.3-0.4 (typically after 1-2 h), take a pre-induction sample of the culture for SDS-PAGE analysis of the overexpression. Calculate the sample size that gives a comparable amount of cells and a good signal on Coomassie stained SDS-PAGE: For a 10 well gel: volume = 0.2 mL/OD600; take half for a 15 well gel.
- For a bacterial culture with an OD600= 0.4, take 500 µL. Take the calculated volume of bacterial culture with a sterile pipette. Spin down the sample (18000 x g, 4 °C, 2 min) and discard the supernatant. Keep the pellet at -20 °C until ready to use for analysis (step 1.11).
- Induce protein expression by pipetting 1.2 mL of a 1 M IPTG stock solution to each 3 L culture solution (final concentration 0.4 mM). Continue incubating the culture at 14 °C for 16 h (overnight).
NOTE: The temperature will typically have reached ~20 °C by the time IPTG is added, depending on the performance of the incubator. - Take a post-induction sample of the culture for SDS-PAGE analysis of the overexpression, following the procedure described in step 1.7.
- Harvest the cells by centrifugation in 1 L tubes (3993 x g, 4 °C, 10 min). Discard the supernatant, keep the cell pellet on ice and proceed directly to the purification.
- Analyze the overexpression by SDS-PAGE35,36. Resuspend the pre- and post-induction samples in 20 µL of running buffer and 20 µL of 2x loading dye. Heat the samples for 5 min at 95 °C in a heat block and load 20 µL on a 15% gel while still hot. Run the gel for 90 min at 180 V. Stain the gel with Coomassie dye according to the instructions of the manufacturer. Compare results with representative results in Figure 1C.
NOTE: The protocol can be stopped here, the cell pellet can be frozen and stored at -80 °C for several weeks. For optimal results, it is recommended to use the fresh bacterial pellet and avoid freezing. Freeze-thaw might lead to lysis of the cells and degradation of Httex1. This might reduce the yield and the quality of the protein.
2. Cell Lysis and Purification of His6-SUMO Httex1 Fusion Protein by Immobilized Metal Affinity Chromatography (IMAC)
- Prepare 2 L of buffer A in a glass bottle (50 mM Tris(hydroxymethyl)-aminomethan (Tris), 500 mM NaCl, 15 mM imidazole). Prepare 1 L of buffer B (50 mM Tris, 500 mM NaCl, 500 mM imidazole pH 7.4) in a glass bottle. After dissolving the salts, adjust the pH with 10 N HCl and filter the solutions into fresh bottles with a bottle top filter (0.65 µm). Prepare a 1000x phenylmethylsulfonyl fluoride (PMSF, 0.3 M) stock solution, aliquot to 100 µL and store at -20 °C.
NOTE: The protocol is designed to enable completing all the steps from lysis of the bacterial pellets to reversed-phase high performance liquid chromatography (RP-HPLC) purification and lyophilization within 8-9 h. To limit aggregation and proteolysis, it is recommended to work rapidly without pausing and perform all steps at 4 °C or on ice. - Add 100 µL of PMSF stock solution and five tablets (1 per 30 mL of final volume) of protease inhibitor to 100 mL of pre-chilled buffer A. Add the bacterial pellet to the buffer and homogenize the suspension by stirring with a magnetic stir bar and by pipetting up and down with a sterile 10 mL pipette (~30 min).
- Divide the bacteria suspension into aliquots of 40 mL in 50 mL disposable plastic tubes. Sonicate each aliquot in a water/ice batch for cell lysis (70% amplitude, total sonication time 5 min, intervals of 30 s sonication, 30 s pause).
NOTE: It is important that the sample does not heat up during the sonication step. It is recommended to add some water to the ice bath to improve heat dissipation during sonication. The sonication procedure might have to be adapted if a different instrument is used. Other lysis methods like a French Press or a microfluidizer should work as well and might be beneficial to avoid heating of the sample and protein aggregation. These devices were not available in our laboratory and we obtained good results with our sonication protocol. - Take a sample of 50 µL of the lysate for sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) analysis. Centrifuge the sample (18000 x g, 4 °C, 2 min) and pipette the soluble fraction in a new test tube. Resuspend the insoluble fraction in 50 µL of buffer A with a pipette. Keep the samples on ice until SDS-PAGE analysis (step 2.6).
- Clarify the lysate by centrifugation (39191 x g, 4 °C, 60 min).
- During the centrifugation step, analyze the cell lysis step by SDS-PAGE. Add 50 µL of 2x loading dye to the soluble and insoluble fraction of the lysate respectively. Heat for 5 min at 95 °C and load 2 µL on a 15% gel while still hot. Run the gel for 90 min at 180 V. Stain the gel with Coomassie dye according to the instructions of the manufacturer. Compare results with representative results in Figure 1C.
- Filter the supernatant (0.45 µm, syringe filters). Take a sample of 20 µL of the filtered supernatant for SDS-PAGE analysis (step 2.11).
NOTE: Typically, a volume of 90 to 100 mL of clarified and filtered supernatant is obtained. Typically, 3 syringe filters are sufficient. If the filtration is cumbersome, try to increase the centrifugation speed and/or time. - Isolate the His6-SUMO Httex1 fusion protein from the clarified bacterial lysate by immobilized metal affinity chromatography (IMAC) on a fast performance liquid chromatography (FPLC) system at 4 °C37.
- Fill the clarified lysate into a superloop and load onto the Ni-NTA column (stripped, cleaned and reloaded according to the manual of the manufacturer, a previous blank run is recommended) at 2 mL/min. Pass 10 column volumes (CV, 200 mL) of buffer A at 10 mL/min to wash out the unbound proteins.
- Elute the fusion protein with 2.5 CV (50 mL) of 100% of buffer B at 2 mL/min. Use a fraction size of 50 mL for loading and washing and 5 mL for elution. Compare results with representative results in Figure 1D.
- Take a sample of each fraction for SDS-PAGE analysis (20 µL) and pool the fractions containing the fusion protein according to the peak of the IMAC chromatogram. Add (2S,3S)-1,4-Bis(sulfanyl)butane-2,3-diol (DTT) and L-cysteine (final concentration 100 mM each) as a powder and dissolve by gently inverting the tube.
NOTE: In our experience the purity of the fusion protein in the different fractions is comparable. As a precaution, the fractions of the purified fusion protein should be pooled quickly after IMAC to prevent aggregation of the highly concentrated fractions. In addition, it is recommended to proceed directly to the cleavage of the SUMO tag and HPLC purification. If necessary, the protocol can be stopped here. The diluted solution of the fusion protein was frozen in liquid nitrogen, stored at -80 °C and purified after thawing without a significant reduction in yield. Storage of the diluted solution of the fusion protein at 4 °C for 24 h also gives similar results. - Analyze the IMAC by SDS-PAGE. Add 20 µL of loading dye to each sample. Load 2 µL of the crude material (2.7), the unbound fraction, the wash fraction and each fraction of the elution peak on a 15% gel. Run the gel for 90 min at 180 V. Stain the gel with Coomassie dye according to the instructions of the manufacturer. Compare results with representative results in Figure 1D.
3. Cleavage of the His6-SUMO-tag and HPLC Purification
Caution: Trifluoroacetic acid (TFA) is a volatile liquid and can cause severe burns so handle with care. Carry out all handling in a fume hood and wear adequate personal protective equipment (i.e., disposable nitrile gloves, safety glasses and a lab coat).
- In a 5 L bottle, add 5 mL of TFA with a plastic syringe to 5 L of water (solvent A: H2O, 0.1% (TFA). Add 2.5 mL of TFA with a plastic syringe to a 2.5 L bottle of acetonitrile (solvent B: acetonitrile, 0.1% TFA).
- Prepare the HPLC system as suggested by the manufacturer. Perform a blank run to ensure a clean column.
- Take a sample of 100 µL of the fusion protein before the addition of ULP1 to monitor the cleavage reaction by UPLC (step 3.5).
- Transfer 20 mL of the fusion protein to a new 50 mL tube and add 0.4 mL of ULP1 stock solution, incubate on ice. Keep the remaining fusion protein on ice.
NOTE: The His-tagged catalytic fragment 403-621 of the Ubiquitin-like-specific protease 1 (here referred to as "ULP1") was used to cleave the SUMO tag. The fusion protein is more stable than the cleaved Httex1. It is recommended not to cleave the SUMO tag of the whole batch. Instead, continue with aliquots of a size that can be directly and completely applied to the HPLC column. - Every 10 minutes, take a sample of 100 µL of the cleavage reaction to monitor the progress by ultra-performance liquid chromatography (UPLC). Centrifuge the samples (18000 rpm, 4 °C, 2 min) and analyze 2 µL of the supernatant by UPLC (gradient from 10% to 90% solvent B in A for 0.25 to 3 min, 10% B for 1 min, refer to the instructions of the manufacturer for instrument usage). Compare the chromatograms obtained for the sample before addition of ULP1 and the samples taken after. Compare the results with representative results in Figure 2B.
- Once the SUMO-cleavage is complete (the peak of the fusion protein has disappeared in the UPLC chromatogram and is fully converted to the newly appearing SUMO and Httex1 peak), filter the sample with a syringe filter (0.22 µm).
NOTE: The SUMO cleavage is typically very fast (10-20 min at 4 °C) therefore UPLC analysis with a run time of 4 min is a valuable tool to monitor the reaction. Filtering the sample before HPLC purification is mainly a preventive measure to increase the lifetime of the column. The sample should not turn turbid. - Purify the filtered sample by RP-HPLC (gradient of 25-35% solvent B in solvent A, over 40 min at 15 mL/min. (0-10 min: 5%; 10-12.5 min: 5 to 25%; 12.5-52.5 min: 25 to 35%; 52.5-57.5 min 35 to 95%; refer to the instructions of the manufacturer for instrument usage). Compare results with representative results in Figure 2C.
NOTE: Httex1 and His6-SUMO separate well by RP-HPLC. However, there can be small amounts of truncated Httex1 in the beginning and end of the peak. Collect small fractions to obtain the maximum amount of pure material.
Caution: Use the appropriate safety equipment (i.e., lab coat, insulated gloves and a face shield) when handling cryogenic fluids. - Analyze the HPLC-fractions by electro-spray ionization mass spectrometry (ESI-MS, autosampler, inject 10 µL, flow 0.6 mL/min, solvent: 20% B in A, no column, refer to the instructions of the manufacturer for instrument usage) and UPLC (gradient from 10% to 90% solvent B in A for 0.25 to 3 min, 10% B for 1 min, refer to the instructions of the manufacturer for instrument usage). Pool fractions of similar purity in 50 mL plastic tubes, freeze in liquid nitrogen and lyophilize. Weigh and transfer the lyophilized protein into 2 mL plastic tubes and store at -20 °C.
- Characterize the purified material by UPLC, ESI-MS and SDS-PAGE. Dissolve 100 µg of lyophilized Httex1 in 8 µL of neat TFA in a 1.5 mL tube and incubate for 20 min at room temperature in the closed test tube. Carefully evaporate the TFA under a fume hood with a stream of nitrogen or argon. Use low pressure of nitrogen/argon to avoid loss of the sample.
- Dissolve the protein in 100 µL of H2O. Analyze 2 µL by UPLC and 5 µL by ESI-MS as in step 3.8. Mix 20 µL of protein solution with 20 µL of 2x loading dye.
- Analyze amounts of 1 µg to 10 µg by SDS-PAGE. Run the gel for 90 min at 180 V. Stain the gel with Coomassie dye according to the instructions of the manufacturer. Compare results with representative results in Figure 2D.
4. Disaggregation and Resolubilization of Httex1 Proteins
Caution: TFA is a volatile liquid and can cause severe burns so handle with care. Carry out all handling in a fume hood and wear adequate personal protective equipment (i.e. disposable nitrile gloves, safety glasses and a lab coat).
- Prepare 10 mL of Dulbecco's phosphate buffered saline (DPBS) (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4, pH 7.4) from the premixed powder in a 50 mL tube. Filter the DPBS solution through a 0.2 µm filter before each use.
- Dissolve 150 µg of lyophilized Httex1 in 12 µL of neat TFA in a 1.5 mL tube and incubate for 20 min at room temperature in the closed test tube. Carefully evaporate the TFA under a fume hood with a stream of nitrogen or argon. Use low pressure of nitrogen/argon to avoid loss of the sample38.
NOTE: In general, use 4 µL TFA to dissolve and disaggregate 50 µg of protein. This procedure will create a film of protein on the inside walls of the test tube. To prevent immediate aggregation of Httex1 in the following steps, work with pre-cooled buffers, keep the protein always on ice and avoid high concentrations. - Dissolve the disaggregated protein in 1 mL of pre-cooled DPBS and adjust the pH to 7.2-7.4 with 1 M NaOH. Filter the protein solution through a 100 kDa centrifugal filter into 1.5 mL plastic tubes (20000 x g, 4 °C, 20 minutes).
NOTE: The calculated theoretical concentration of Httex1 is higher than the desired final concentration to account for possible loss. The filtration step is necessary to remove any aggregates that might have formed during dissolution of the protein. - Determine the concentration of Httex1 using a UPLC calibration curve based on amino acid analysis (detection at λ214) and send 2 µg of the protein to amino acid analysis to validate the concentration10. Calculate the amount of DPBS that needs to be added to obtain a concentration of 3 µM Httex1.
- Dilute the protein to 3 µM by adding the calculated amount of DPBS to the test tube. Keep the tube on ice until the initiation of the aggregation protocol.
NOTE: Httex1 43Q should not be stored in solution. Always prepare a fresh protein solution based on the protocol above.Httex1 proteins are best stored as a lyophilized powder at -20°C.
5. Monitoring of the Aggregation Kinetics of Httex1 43Q using UPLC and circular Dichroism (CD) Spectroscopy and Characterization of the Aggregates by Transmission Electron Microscopy (TEM)
- Prepare uranyl formate solution for TEM as previously reported39.
- Initiate the aggregation of Httex1 43Q by incubating a 3 µM solution in DPBS at 37 °C (use 1 mL of solution prepared as described above in the disaggregation protocol).
NOTE: The aggregation of Httex1 can be performed at higher concentrations depending on the needs and aims of the experiment. - Quantify the amount of soluble protein using UPLC at indicated time points (at 0, 1, 2, 4, 6, 8, 12, 24, 48 and 120 h). To do that, take an aliquot of 35 µL and remove the insoluble aggregates by centrifugation (20000 x g, 4 °C, 20 min). Inject 4 µL of the supernatant into the UPLC. Calculate the proportion of soluble monomer based on the change of the peak area using the instrument software 40. Compare results with representative results in Figure 3A.
- Characterize the changes in secondary structure using CD spectroscopy at 0 and 48 h. Take an aliquot of 100 µL and measure the ellipticity (1 mm quartz cuvette, 195 nm to 250 nm, 20 °C, data points every 0.2 nm, speed 10 nm/min, digital integration time 2 s, bandwidth of 1.0 nm). Acquire 6 spectra of the sample and average and smooth using a binomial filter with convolution width of 99. Plot the spectra as the mean residue molar ellipticity (θMRE)41. Compare results with representative results in Figure 3B.
- Characterize the structural and morphological properties of the aggregates by TEM. Put 3 µL of the protein solution onto a Formvar/carbon-coated 200-mesh, glow-discharged copper grid for 1 min. Wash the grid twice with 15 μL water, once with 15 μL of 0.7% (w/v) uranyl formate and stain for 30 s with 15 μL of 0.7% w/v uranyl formate. Perform a TEM analysis of the grids. Compare results with representative results in Figure 3C.
Representative Results
Httex1 is expressed in E. coli with an N-terminal His6-SUMO tag. The representative results of the expression and purification of the fusion protein are summarized in Figure 1. The sequence of Httex1 consists of the residues 2-90 of Htt and starts with Ala2, because Met1 is fully cleaved in vivo42. The numbering of the amino acids refers to the 23Q variant, the complete sequence of the expressed fusion protein is shown in Figure 1A. The plasmids will be deposited at Addgene in the near future to be shared with the community. A schematic of the plasmid and the expressed fusion protein is shown in Figure 1B. His6-SUMO Httex1 expresses at a medium level (Figure 1C) and most of the fusion protein is present in the soluble fraction after lysis, both for the 23Q and the 43Q variant. The fusion protein migrates higher than expected, based on the molecular weight. This is partly due to the strong fold of SUMO but mostly due to the unusual sequence composition of Httex1, containing mainly glutamine and proline residues. Both the wildtype (23Q) and the mutant (43Q) fusion protein can be enriched to ~80% purity by IMAC (Figure 1D) The presence of co-purifying host protein can be explained by the comparatively low expression level of Httex1 and the big sample volume applied to the column.
The cleavage of the His6-SUMO tag and the purification of Httex1 is shown in Figure 2A. UPLC is an efficient tool to monitor the cleavage of the His6-SUMO tag (Figure 2B). The original peak of the fusion protein is consumed and two new and well-separated peaks corresponding the His6-SUMO tag and Httex1 appear. The cleavage reaction is finished in 10-20 min. The Western Blot (WB) is too slow to monitor the cleavage reaction efficiently, but it has been included in the figure for reference and to demonstrate the completeness of the SUMO cleavage. Both Httex1 23Q and 43Q can be separated well from the His6-SUMO tag by RP-HPLC (Figure 2C) and were obtained in high purity as shown by UPLC, MS and SDS-PAGE analysis (Figure 2D).
To illustrate that the Httex1 proteins prepared by this method retain the expected aggregation properties of Httex1, we assessed the fibrillization kinetics of mutant Httex1 at 37 °C by a sedimentation assay, monitored the changes in secondary structure by CD spectroscopy, and characterized the morphology of the aggregates by TEM. A representative data set of the aggregation kinetics of mHttex1 fibril formation as determined by a sedimentation assay is shown inFigure 3A. The loss of soluble Httex1 43Q over time, due to fibril formation was quantified by UPLC. We observe a complete depletion of soluble protein after 48 hours of incubation. Additionally, we determined the secondary structure of the protein by CD spectroscopy (Figure 3B). Httex1 43Q shifts from unstructured (λmin 205 nm) to mainly β-sheet rich conformation (λmin 215 nm) after 48 hours of incubation. This structural change is accompanied by the formation of long fibrillar aggregates as observable by TEM at 48 hours (Figure 3C).
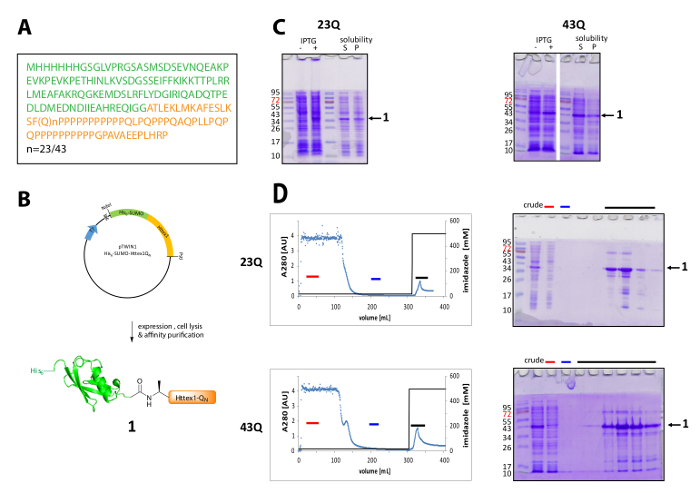
Figure 1. Expression and purification of the His6-SUMO Httex1 fusion protein.
(A) The amino acid sequence of the His6-SUMO-Httex1-QN fusion constructs (His6-SUMO in green and Httex1-QN in orange); (B) Schematic overview of the expression and purification of the fusion protein; (C) SDS-PAGE analysis of the expression and the solubility of the fusion protein after lysis; (D) Chromatogram of the IMAC purification of the fusion protein and analysis of the fractions by SDS-PAGE (red bar: unbound fraction, blue bar: wash fraction, black bar: fractions containing the elution peak); Please click here to view a larger version of this figure.
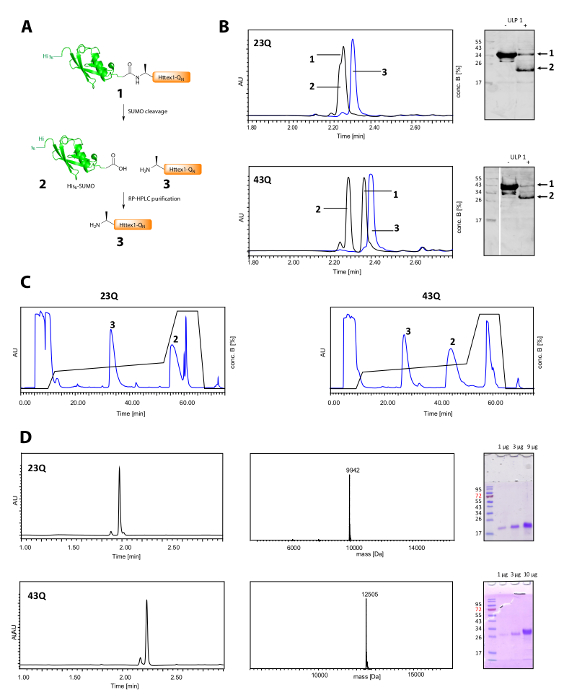
Figure 2. Cleavage of the His6 SUMO tag and purification of tag-free Httex1-QN proteins.
(A) schematic overview; (B) analysis of the cleavage of the SUMO tag with ULP1 by UPLC (blue: before addition of ULP1; black: 20 min (23Q), respectively 10 min (43Q) after addition of ULP1) and WB (MAB5492 1:2000, secondary goat anti mouse antibody 1:5000); (C) Chromatogram of the preparative RP-HPLC purification of Httex1; D: analysis of the purified Httex1 by UPLC, SDS-PAGE and ESI-MS; the expected molecular weight is 9943 Da (23Q) and 12506 Da (43Q) respectively. Please click here to view a larger version of this figure.
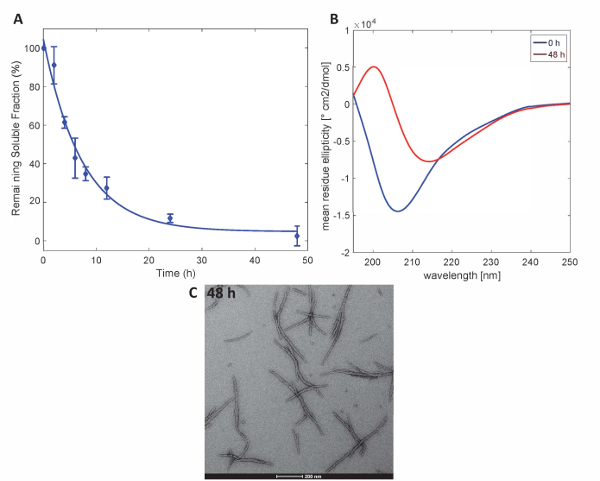
Figure 3. Aggregation of Httex1-43Q: (A) Sedimentation assay based on UPLC. (B) CD spectra of the secondary structure at 0 h and 48 h. (C) TEM micrographs of the aggregates at 48 h (scale bars are 200 nm). Please click here to view a larger version of this figure.
Discussion
In this protocol, we have outlined an efficient procedure for obtaining milligram quantities of native, untagged Httex1 containing 23 or 43 glutamine residues. This was achieved by expressing Httex1 as a C-terminal fusion to a His6-SUMO tag, which is used to isolate the fusion protein from the cell lysate by IMAC and is cleaved prior to HPLC purification of Httex1. While the SUMO strategy has been used in the production of several other proteins, our method shows that the unique properties SUMO could also be used to generate intrinsically disordered, aggregation-prone, amyloidogenic protein that have previously proved to be extremely difficult to handle and produce43,44. We present a protocol that is straightforward, easy to use and comparable to a protocol for the generation of a "well-behaved" protein. The SUMO fusion solubilizes and stabilizes Httex1 during expression and the IMAC purification step. Premature cleavage of the tag, as observed with the intein strategy10 and aggregation were no longer an issue.
Intrinsically disordered proteins are especially vulnerable to degradation. While N-terminal degradation in the N17 region is not an issue using this protocol, truncations in the PRD of Httex1 can occur. As the truncated proteins are very similar to Httex1 in hydrophobicity, charge and size, removing them by chromatographic means is challenging, thus it is best to prevent their formation in the first place. Sticking closely to the protocol, always working on ice and using a sufficient amount of protease inhibitor should help keep the level of observed truncation very low. Applying a fusion tag at the C-terminus of Httex1 could remove truncations in the PRD easily as the truncated protein would lose the affinity tag as well. However, if the native sequence needs to be maintained this option cannot be applied as Httex1 ends with proline and to the best of our knowledge there are no C-terminal fusion tags that are known to induce traceless and efficient cleavage after proline.
The most critical part of the protocol is the handling of the Httex1 liberated after cleavage of the SUMO tag by ULP1. The protein should be purified immediately by RP-HPLC. Fortunately, this is an efficient and fast reaction that is usually completed in 10-20 min at 4 °C. In contrast, the intein strategy required several hours for complete cleavage of the intein, thus requiring a trade-off between incomplete cleavage and beginning aggregation in order to maximize the yield. A fast workup is required for mutant Httex1, as it will start to aggregate at the comparatively high concentration present in the cleavage reaction, whereas the 23Q variant is stable for a longer time. During the RP-HPLC purification, another advantage of SUMO becomes apparent: While the Ssp DnaB intein is hydrophobic and sticks strongly to the column, SUMO is more hydrophilic and elutes completely from the C4 reversed-phase column. Although commercial ULP1 is quite costly, the protein can be easily produced in high yield following previously published protocols29.
The critical importance of applying a disaggregation protocol prior to using Httex1 cannot be stressed enough. Lyophilized polyQ proteins such as Httex1 are stable and can be stored long periods, but are not completely soluble in water and buffers. The presence of preformed oligomers or fibrils could have a significant impact on aggregation kinetics and biophysical properties of the protein45. The disaggregation protocol described here allows the disaggregation of the protein, removal of preformed aggregates and generation of a solution of monomeric Httex1 from a lyophilized sample. We observed similar aggregation kinetics and fibril morphology for Httex1 obtained with the SUMO and the intein strategy.
Compared to previous methods for producing Httex1, the SUMO strategy described here offers several advantages and expands the range of possible studies to investigate the structure and functional properties of this protein in health and disease. The SUMO-Httex1 fusion protein is easy to handle, it can be frozen and stored or kept in solution for 24 h at ambient temperature, while the free mHttex1 would aggregate quickly. The stability and high solubility of the SUMO-Httex1 fusion proteins provide greater flexibility to manipulate the protein and/or introduce enzymatic and chemical modifications into mHttex1 that would otherwise not be possible after cleavage. This includes the introduction of post-translational modifications, fluorophores, spin labels, biotin tags, etc. The advances presented here should 1) facilitate future studies to elucidate structure-function relationships of Httex1; 2) generate new tools to investigate Htt aggregation and pathology spreading; 3) enable the development of new assays to identify molecules that stabilize mutant Httex1 and prevent its aggregation; and 4) encourage scientists from other fields to bring to work on this protein and join our quest to find cures for Huntington's disease.
Disclosures
The authors have nothing to disclose.
Acknowledgements
This work was funded primarily by grants from the CHDI foundation and the Swiss National Science Foundation. We thank Dr. Sophie Vieweg for useful discussions during the development of this new expression system and other members of the Lashuel group for sharing their experience with this expression system and for their input and valuable feedback. We also thank Prof. Oliver Hantschel for providing the ULP1 plasmid. The authors thank Dr. John B. Warner and Dr. Senthil K. Thangaraj for critical review of the manuscript
Materials
| Uranyl formate (UO2(CHO2)2) | EMS | 22450 | |
| Formvar/carbon 200 mesh, Cu 50 grids | EMS | FCF200-Cu-50 | |
| High Precision Cell made of Quartz SUPRASIL 1 mm light path from Hellma Analytics | HellmaAnalytics | 110-1-40 | |
| Buffer Substance Dulbecco's (PBS w/o Ca and Mg) ancinne ref 47302 (RT) SERVA | Witech | SVA4730203 | |
| Ampicillin | AxonLab | A0839.0100 | |
| Luria Broth (Miller's LB Broth) | Chemie Brunschwig | 1551.05 | |
| Isopropyl β-D-1-thiogalactopyranoside (IPTG) | AxonLab | A1008.0025 | |
| E. coli B ER2566 | NEB | NEB# E4130 | |
| Imidazole | Sigma | 56750-500G | |
| cOmplete Protease Inhibitor Cocktail | Roche | 4693116001 | |
| Anti-Huntingtin Antibody, a.a. 1-82 | Merck Millipore Corporation | MAB5492 | |
| IRDye 680RD Goat anti-Mouse IgG (H + L) | Licor | 925-68070 | |
| PMSF | AxonLab | A0999.0005 | |
| HisPrep 16/10 column | GE Healthcare | 28936551 | |
| C4 HPLC column | Phenomenex | 00G-4168.P0 | 10 µm C4 300 Å, LC Column 250 x 21.2 mm, Phenomenex, 19×10 mm guard column, not temperature jacketed |
| Acetonitrile HPLC | MachereyNagel | C2502 | |
| Filtre seringue Filtropur S 0,45 ul sans prefiltre sterile | Sarstedt AG | 83.1826 | |
| Spectrophotometer semi-micro cuvette | Reactolab S.A. | 2534 | |
| Superloop, 1/16" fittings (ÄKTAdesign), 50 ml | GE Healthcare | 18111382 | |
| Trifluoroacetic acid | Sigma | 302031 | |
| GREINER Tubes fo FPLC 16 x 100 mm, cap. 12.0 ml | Greiner Bio-One | 7.160 102 | |
| 100 kD Microcon fast flow filters | Merck Millipore Corporation | MRCF0R100 | |
| Vibra-cell VCX130 ultrasonic liquid processor | Sonics | ||
| Äkta 900 equipped with a fraction collector | GE Healthcare | ||
| Jasco J-815 Circular Dichroism | Jasco | ||
| Waters UPLC system | Waters | C8 BEH acquity 2.1×100 mm 1.7 micron column , preheated column (40 °C), flow rate of 0.6 mL/min, injection volume of 4 μL | |
| waters HPLC system | Waters | 2489 UV detector and 2535 quaternary gradient module, 20 mL loop in a FlexInject housing | |
| ESI-MS: Finnigan LTQ | Thermo Fisher Scientific | ||
| lyophylizer instrument | FreeZone 2.5 Plus | ||
| Tecnai Spirit BioTWIN | FEI | electron microscope equipped with a LaB6 gun and a 4K x 4K FEI Eagle CCD camera (FEI) and operated at 80 kV | |
| 37 °C shaking incubator | Infors HT multitron Standard | ||
| Biophotometer plus | Eppendorf |
References
- Saudou, F., Humbert, S. The Biology of Huntingtin. Neuron. 89 (5), 910-926 (2016).
- MacDonald, M. E., Gines, S., Gusella, J. F., Wheeler, V. C. Huntington’s disease. Neuromolecular Medicine. 4 (1-2), 7-20 (2003).
- Li, S., Li, X. J. Multiple pathways contribute to the pathogenesis of Huntington disease. Molecular Neurodegeneration. 1, 19 (2006).
- DiFiglia, M. Aggregation of Huntingtin in Neuronal Intranuclear Inclusions and Dystrophic Neurites in Brain. Science. 277 (5334), 1990-1993 (1997).
- Atwal, R. S., et al. Huntingtin has a membrane association signal that can modulate huntingtin aggregation, nuclear entry and toxicity. Human Molecular Genetics. 16 (21), 2600-2615 (2007).
- Sathasivam, K., et al. Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proceedings of the National Academy of Sciences U S A. 110 (6), 2366-2370 (2013).
- Mangiarini, L., et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 87 (3), 493-506 (1996).
- El-Daher, M. T., et al. Huntingtin proteolysis releases non-polyQ fragments that cause toxicity through dynamin 1 dysregulation. EMBO Journal. 34 (17), 2255-2271 (2015).
- Lunkes, A., et al. Proteases acting on mutant Huntingtin generate cleaved products that differentially build up cytoplasmic and nuclear inclusions. Molecular Cell. 10 (2), 259-269 (2002).
- Vieweg, S., Ansaloni, A., Wang, Z. M., Warner, J. B., Lashuel, H. A. An Intein-based Strategy for the Production of Tag-free Huntingtin Exon 1 Proteins Enables New Insights into the Polyglutamine Dependence of Httex1 Aggregation and Fibril Formation. Journal of Biological Chemistry. 291 (23), 12074-12086 (2016).
- Georgalis, Y., et al. Huntingtin aggregation monitored by dynamic light scattering. Proceedings of the National Academy of Sciences U S A. 95 (11), 6118-6121 (1998).
- Scherzinger, E., et al. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell. 90 (3), 549-558 (1997).
- Scherzinger, E., et al. Self-assembly of polyglutamine-containing huntingtin fragments into amyloid-like fibrils: implications for Huntington’s disease pathology. Proceedings of the National Academy of Sciences U S A. 96 (8), 4604-4609 (1999).
- Muchowski, P. J., et al. Hsp70 and hsp40 chaperones can inhibit self-assembly of polyglutamine proteins into amyloid-like fibrils. Proceedings of the National Academy of Sciences U S A. 97 (14), 7841-7846 (2000).
- Heiser, V., et al. Inhibition of huntingtin fibrillogenesis by specific antibodies and small molecules: implications for Huntington’s disease therapy. Proceedings of the National Academy of Sciences U S A. 97 (12), 6739-6744 (2000).
- Bennett, E. J., Bence, N. F., Jayakumar, R., Kopito, R. R. Global impairment of the ubiquitin-proteasome system by nuclear or cytoplasmic protein aggregates precedes inclusion body formation. Molecular Cell. 17 (3), 351-365 (2005).
- Tam, S., et al. The chaperonin TRiC blocks a huntingtin sequence element that promotes the conformational switch to aggregation. Nature Structural & Molecular Biology. 16 (12), 1279-1285 (2009).
- Nekooki-Machida, Y., et al. Distinct conformations of in vitro and in vivo amyloids of huntingtin-exon1 show different cytotoxicity. Proceedings of the National Academy of Sciences U S A. 106 (24), 9679-9684 (2009).
- Wacker, J. L., Zareie, M. H., Fong, H., Sarikaya, M., Muchowski, P. J. Hsp70 and Hsp40 attenuate formation of spherical and annular polyglutamine oligomers by partitioning monomer. Nature Structural & Molecular Biology. 11 (12), 1215-1222 (2004).
- Legleiter, J., et al. Monoclonal antibodies recognize distinct conformational epitopes formed by polyglutamine in a mutant huntingtin fragment. Journal of Biological Chemistry. 284 (32), 21647-21658 (2009).
- Legleiter, J., et al. Mutant huntingtin fragments form oligomers in a polyglutamine length-dependent manner in vitro and in vivo. Journal of Biological Chemistry. 285 (19), 14777-14790 (2010).
- Nucifora, L. G., et al. Identification of novel potentially toxic oligomers formed in vitro. from mammalian-derived expanded huntingtin exon-1 protein. Journal of Biological Chemistry. 287 (19), 16017-16028 (2012).
- Dahlgren, P. R., et al. Atomic force microscopy analysis of the Huntington protein nanofibril formation. Nanomedicine. 1 (1), 52-57 (2005).
- Poirier, M. A., et al. Huntingtin spheroids and protofibrils as precursors in polyglutamine fibrilization. Journal of Biological Chemistry. 277 (43), 41032-41037 (2002).
- Duim, W. C., Chen, B., Frydman, J., Moerner, W. E. Sub-diffraction imaging of huntingtin protein aggregates by fluorescence blink-microscopy and atomic force microscopy. Chemphyschem. 12 (13), 2387-2390 (2011).
- Pieri, L., Madiona, K., Bousset, L., Melki, R. Fibrillar alpha-synuclein and huntingtin exon 1 assemblies are toxic to the cells. Biophysical Journal. 102 (12), 2894-2905 (2012).
- Monsellier, E., Redeker, V., Ruiz-Arlandis, G., Bousset, L., Melki, R. Molecular interaction between the chaperone Hsc70 and the N-terminal flank of huntingtin exon 1 modulates aggregation. Journal of Biological Chemistry. 290 (5), 2560-2576 (2015).
- Isas, J. M., Langen, R., Siemer, A. B. Solid-State Nuclear Magnetic Resonance on the Static and Dynamic Domains of Huntingtin Exon-1 Fibrils. 생화학. 54 (25), 3942-3949 (2015).
- Malakhov, M. P., et al. SUMO fusions and SUMO-specific protease for efficient expression and purification of proteins. Journal of Structural Function Genomics. 5 (1-2), 75-86 (2004).
- Mossessova, E., Lima, C. D. Ulp1-SUMO crystal structure and genetic analysis reveal conserved interactions and a regulatory element essential for cell growth in yeast. Molecular Cell. 5 (5), 865-876 (2000).
- Kumari, S., Pal, R. K., Gupta, R., Goel, M. High Resolution X-ray Diffraction Dataset for Bacillus licheniformis Gamma Glutamyl Transpeptidase-acivicin complex: SUMO-Tag Renders High Expression and Solubility. Protein Journakl. 36 (1), 7-16 (2017).
- Zhang, J., Sun, A., Dong, Y., Wei, D. Recombinant Production and Characterization of SAC, the Core Domain of Par-4, by SUMO Fusion System. Applied Biochemistry and Biotechnology. , (2017).
- Reif, A., et al. Semisynthesis of biologically active glycoforms of the human cytokine interleukin 6. Angewandte Chemie International Edition English. 53 (45), 12125-12131 (2014).
- Froger, A., Hall, J. E. Transformation of plasmid DNA into E. coli using the heat shock method. Journal of Visualized Experiments. (6), 253 (2007).
- Smith, B. J. SDS Polyacrylamide Gel Electrophoresis of Proteins. Methods in Molecular Biology. 1, 41-55 (1984).
- Lawrence, A. M., Besir, H. U. Staining of proteins in gels with Coomassie G-250 without organic solvent and acetic acid. Journal of Visualized Experiments. (30), (2009).
- Block, H., et al. Immobilized-metal affinity chromatography (IMAC): a review. Methods in Enzymology. 463, 439-473 (2009).
- Chen, S. M., Wetzel, R. Solubilization and disaggregation of polyglutamine peptides. Protein Science. 10 (4), 887-891 (2001).
- Booth, D. S., Avila-Sakar, A., Cheng, Y. Visualizing proteins and macromolecular complexes by negative stain EM: from grid preparation to image acquisition. Journal of Visualized Experiments. (58), (2011).
- O’Nuallain, B., et al. Kinetics and thermodynamics of amyloid assembly using a high-performance liquid chromatography-based sedimentation assay. Amyloid, Prions, and Other Protein Aggregates, Pt C. 413, 34-74 (2006).
- Greenfield, N. J. Analysis of circular dichroism data. Methods in Enzymology. 383, 282-317 (2004).
- Aiken, C. T., et al. Phosphorylation of threonine 3: implications for Huntingtin aggregation and neurotoxicity. Journal of Biological Chemistry. 284 (43), 29427-29436 (2009).
- Satakarni, M., Curtis, R. Production of recombinant peptides as fusions with SUMO. Protein Expression and Purification. 78 (2), 113-119 (2011).
- Davies, H. A., Wilkinson, M. C., Gibson, R. P., Middleton, D. A. Expression and purification of the aortic amyloid polypeptide medin. Protein Expression and Purification. 98, 32-37 (2014).
- Chen, S., Wetzel, R. Solubilization and disaggregation of polyglutamine peptides. Protein Science. 10 (4), 887-891 (2001).
