Stentor cultures have been reliably established and maintained from individual cells or cell fragments using Section 1 of the protocol. A representative example of a large healthy culture is shown in Video 1.
The time course of regeneration was measured in Stentor using the sucrose treatment method for initiating regeneration outlined in Step 3.1, combined with the imaging and analysis method discussed in Section 4 (Figure 7). This plot indicates that there is a one-hour-long spread in the time taken by the population of cells to reach any particular stage. This type of analysis allows the study of temporal heterogeneity in the regeneration process in a population of regenerating cells.
The following is a summary of regeneration timing that has been observed thus far after dozens of sucrose treatments (Figure 4 and Figure 6). Stage 1 is when Stentor cells look like teardrops without any membranellar band (this stage starts immediately after sucrose washout). This stage lasts for 3 – 6 h. Stage 2 is when a membranellar band appears and grows (3 – 6 h after sucrose treatment). This stage lasts for 3 – 4 h. Stage 3 is when an oral primordium appears at the posterior end of the membranellar band (6 – 8 h after sucrose treatment), and both structures are moved toward the anterior end of the cell. This stage lasts for 1 – 2 h. When both the membranellar band and the oral apparatus reached the anterior end of the cell, this indicated the completion of regeneration. The cell has adopted characteristic Stentor trumpet-like shape. Cells were completely regenerated 8 – 9 h after sucrose treatment.
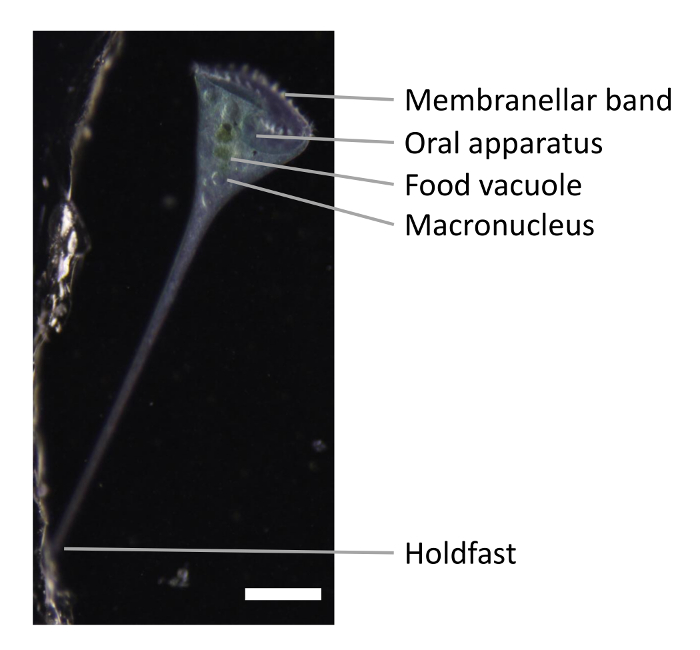
Figure 1. Snapshot of Stentor. The membranellar band and the oral apparatus are shown at the anterior end of the cell. The holdfast is at the posterior end of the cell. Stentor macronucleus is nodulated. A well-fed Stentor has green food vacuoles containing mostly Chlamydomonas. Scale bar is 0.5 mm. Please click here to view a larger version of this figure.

Figure 2. Stentor cutting set-up. Cell cutting is performed by manually manipulating a glass needle while looking at the cell using a commercially available dissecting stereo microscope. The purpose of the tissue paper is to provide the white background to see the cells better. Please click here to view a larger version of this figure.
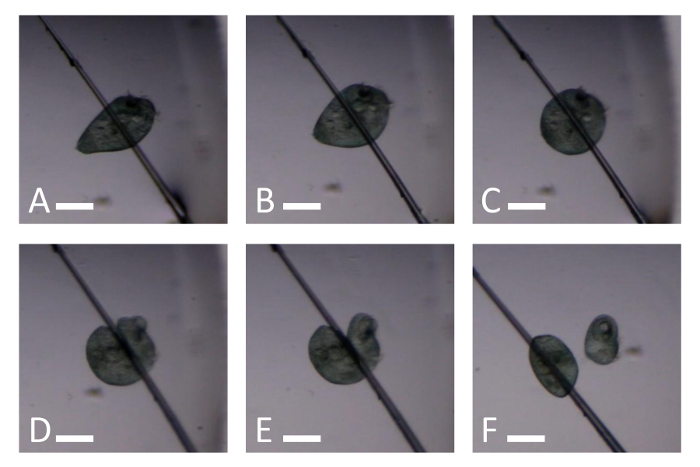
Figure 3. Snapshots of Video 3 illustrating how to cut a Stentor with a glass needle. (A) A Stentor immediately before the needle touches it. (B) A Stentor gently squeezed between a needle and glass slide. (C) A contracted Stentor reacting to the force of the needle. (D) A Stentor being cut by gently pressing on the cell with the side of the needle. (E) A Stentor now cut in two but not separated. (F) Two Stentor fragments. Scale bar is 0.25 mm. Please click here to view a larger version of this figure.
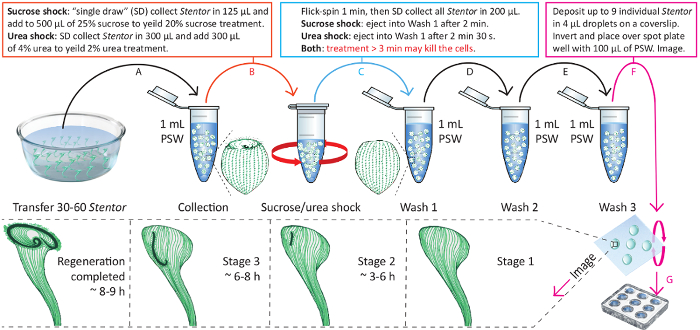
Figure 4. Schematic visualization of Section 3 and Section 4 of the protocol. This is an illustrated protocol for performing sucrose or urea treatment and observation of cell regeneration. The time indicated in the bottom panel is measured from the beginning of the Wash 1. Please click here to view a larger version of this figure.

Figure 5. The setup for imaging and/or direct observation of regeneration with an upright microscope. In each 4 µL droplet hanging from the coverslip, there is one cell undergoing regeneration. Water in the wells of the glass spot plate limits evaporation of the droplets. This setup allows following the regeneration of multiple cells in parallel. Please click here to view a larger version of this figure.

Figure 6. Snapshots of Stentor in each stage of the membranellar band and the oral apparatus regeneration. (A) Stage 1 is characterized by a teardrop-like cell shape and the absence of the membranellar band. (B) Stage 2 is characterized by the appearance of the membranellar band, a cilia-based structure that beats continuously. Membranellar bands are marked with white dashed lines in panels B – D. (C and D) Stage 3 is characterized by the appearance of oral primordium (marked with an arrow). Oral primordium looks like an invagination at the posterior end of the membranellar band. The oral primordium will then move up towards the anterior of the cell to become the oral apparatus. (E) Regeneration is completed when the cell has adopted the characteristic Stentor trumpet-like shape, and the oral apparatus (marked with an arrow) has widened at the anterior end of the cell. Scale bar is 0.25 mm. Please click here to view a larger version of this figure.
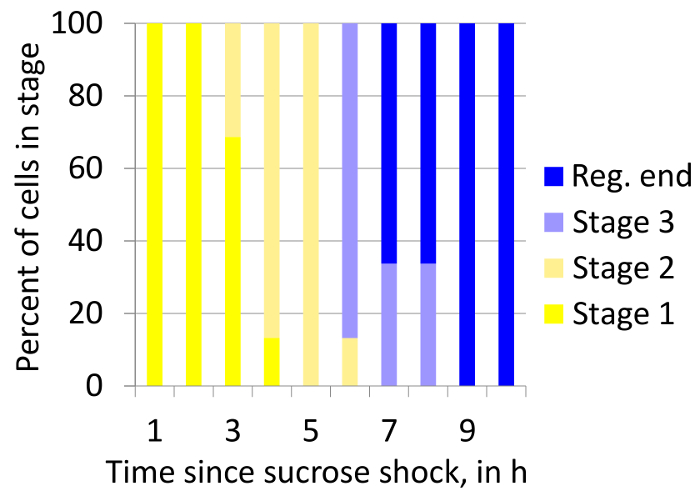
Figure 7. Stacked box plot showing how proportions of cells in all stages of regeneration after sucrose treatment changes over over time. The plot shows heterogeneity in the timing of regeneration stages within a population of regenerating cells. "Reg. end" indicates the completion of regeneration. The number of cells: 28. Please click here to view a larger version of this figure.

Video 1. Example of a healthy Stentor culture. Generally, if a culture is this concentrated, splitting the culture is recommended. Please click here to view this video. (Right-click to download.)

Video 2. Slowing down Stentor with methylcellulose. Please click here to view this video. (Right-click to download.)

Video 3. Cutting Stentor. Individual cells can be manually cut into multiple pieces under a stereo dissecting microscope by pressing a glass needle through the cell. Please click here to view this video. (Right-click to download.)
| Problem with culture | Possible remedy | Possible prevention |
| Culture is overwhelmed by rotifers or other large organisms. | Remove as many Stentor as possible into a new culture dish. Then wash the cells three times. | Wash Stentor thoroughly when receiving them, even when purchasing them from a commercial supplier. |
| Culture is overwhelmed by fungus or other small organisms. | Identify a corner where there are the least Stentor. In that area, remove as much media as possible without removing many Stentor and add in new PSW. Mix gently but throughly to distrubute the invading organisms. Skip the next feeding and the Stentor will eat them. | Observe the culture regularly and remove small contaminants by exchanging media for fresh PSW. |
| Stentor are laying on top of each other. | Split the culture so that the concentration is less than 20 cells/mL. | Split cultures more often. |
| Stentor cells are mating. | Feed every 4 days instead of every 5 days. | |
| Culture has a lot of dark waste material. | Remove as much of the dark material, Stentor waste, as possible without removing the cells. If Stentor are attached to the dark material, pipette up and down to release the Stentor cells from their waste and then remove the media with the waste without removing the cells. Or, remove those Stentor and feed accordingly less. | Feed Stentor 1 mL less food per 100 mL of culture. |
| Unhealthy-looking (vacuolated/bloated or rounded) Stentor. | Remove as much media as possible without removing many Stentor cells and add new PSW. | Exchange the media regularly. |
Table 1. Troubleshooting guide for culturing Stentor.
