1. Sample Preparation for Native MS of Protein and Protein-Ligand Complexes
NOTE: To gain an understanding of the molecular basis of a protein complex and ligand binding using native MS, suitable sample preparation is key. The aim of this section is to highlight the essential sample preparation steps prior to MS analysis using the HerA-NurA complex which binds DNA and nucleotides as an example.
- Prepare 20 µL aliquots of concentrated purified protein (typically 15 – 30 µM) in a 1.5 mL tube.
- For ATP or ADP binding analysis
NOTE: Add increasing concentrations of the non-hydrolyzable ATP analog adenosine 5′-O-(3-thiotriphosphate), tetralithium salt (ATP-γ-S) or adenosine 5′-diphosphate (ADP). Non-hydrolyzable ATP derivatives generate a stable complex which would enable the ATP-bound protein to be captured. Other non-hydrolysable ATP analogues that could be tested include AMP-PNP and ATP-γ-S-Mg2+.- For HerA-NurA studies, mix 5 µM of purified protein with ATP-γ-S and ADP at concentrations ranging from 0-1 mM.
- To capture simultaneous ATP-γ-S and ADP binding, add both nucleotides at the same or varying concentrations.
- Add 2 mM MgCl2 and incubate at 25 °C in a dry bath incubator for 1 h.
NOTE: Analysis of nucleotide binding using nESI native MS can result in artefactual binding at high concentrations, therefore non-specific binding must be taken into account29. To investigate non-specific binding, add a higher concentration of nucleotides between 2-5 mM).
- For DNA binding analysis
- Mix the protein and DNA at a molar ratio that allows for protein-DNA complex formation. For HerA and HerA-NurA, mix 5 µM of purified protein with DNA at a 1:1 ratio.
- Incubate the HerA-NurA or HerA – DNA mixture for 30 min at 25 °C in a dry bath incubator until it reaches equilibrium. The duration and temperature of incubation may vary depending on the protein under investigation.
- Buffer exchange protein samples to MS compatible buffers. Commonly, aqueous ammonium acetate solution between 5 mM-1 M at pH 7-8 are used. Other MS compatible buffers include ethylenediammonium diacetate (EDDA) and Triethylammonium acetate (TEAA)30. For HerA-NurA studies, use 200 mM ammonium acetate pH 7.
NOTE: There are several methods for buffer exchange prior to analysis by MS such as using a spin concentrator or chromatography columns. Native MS is mostly limited by the quality of the sample such as buffers and adducts used during purification. Therefore, it is essential to perform sufficient desalting to obtain resolved peaks. - For HerA-NurA ligand binding studies, buffer exchange samples 6-8 times into 200 mM ammonium acetate using a concentrator.Although this method is more time consuming, it ensures that resolved peaks are achieved and allows for accurate mass determination of ATP/ADP bound species.
2. Native MS Acquisition and Analysis for Investigating Protein Complexes and Protein-Ligand Complexes
Note: MS conditions should be optimized to achieve highly resolved peaks to enable accurate mass measurements. This section details optimized parameters on a Q-ToF mass spectrometer with a 32k upper limit m/z quadrupole.
- Prepare in-house capillaries for nano-electrospray and perform instrument mass calibration for accurate mass measurements as detailed by Kirshenbaum et al.1.
- Select sensitivity, positive ion acquisition and mobility TOF modes.
- Turn on the Trap, API and IMS gases. For IM separation, use nitrogen (60 mL/min) and argon (8.4 mL/min for the trap region) as starting points and then adjust.
- Set an appropriate m/z acquisition range. For an unknown protein, initial optimization steps should use a wide range such as 500-32,000 m/z.
- Load 2-3 µL of the protein complex solution to be analyzed into a gold coated capillary and insert it into a capillary holder.
- Gently tighten the capillary and place the capillary in the electrospray source stage and slide the stage into position to start acquiring data.
- Apply low nano-flow gas pressure (0.00-0.05 Bar) until a drop is formed at the tip of the capillary. The nano-flow pressure can then be dropped until the spray is maintained.
- Adjust the capillary with respect to the cone by moving the capillary in x, y, z positions and monitor the ion current to achieve a stable ion current. Apply a capillary voltage in the range of 0.9-1.6 kV.
- Set the sampling cone (50-120 V), source offset (60.0), source temperature (25 °C) and cone gas flow (0.0 L/h). These suggested initial conditions can be adjusted.
- To acquire a well resolved mass spectrum and to maximize ion transmission, adjust MS parameters and monitor the resulting change in the spectra. These include adjusting the gas flow in the Trap (2-8 mL/min), He Cell (180 mL/min) and IMS cell (90 mL/min) to achieve best separation at maximum transmission.
- Adjust the trap collision energies if voltage offsets are insufficient. An optimal starting point is between 10-50 V.
NOTE: Increasing the trap energy can remove non-covalently bound adducts. However, take care to avoid collision induced dissociation and unfolding of the protein-ligand complex. Perform ion mobility measurements to check if instrument conditions retain the protein in the native folded state (Step 3). - Improve desolvation by optimizing the trap bias voltage. An optimal starting point is 20-45 V.
- Optimize the wave velocity and wave height to achieve best mobility separation. A detailed explanation and protocol can be found here31. For the HerA-NurA studies, use wave velocity of 40 (m/s) and wave height of 550-650 (V).
- Use all other parameters as instrument default values.
- Prepare a ligand-free sample for analysis as a control for each run (Figure 1). For ligand binding experiments, perform at least three independent measurements.
- Use the Masslynx software to measure masses of generated species and identify the ligand binding, such as ATP and ADP binding and oligomeric states (Figure 2 and 3).Other software available include UniDec32, PULSAR33 and Amphitrite34.
- To quantify the relative abundance of species, use the corresponding ion intensities observed in the raw ESI-MS spectra (for example ligand bound, different oligomers, etc.). Alternatively, perform quantification using specialized software like UniDec and Massign35 (Figure 1 and 3 ).
3. Acquiring and Analyzing IM-MS
NOTE: IM-MS separates ions in the gas-phase based on their size (mass), shape and charge. Every feature resolved in m/z spectrum is associated with a drift time distribution. IM-MS measures the drift-time of an ion which can be used to calculate the collision cross section (CCS). Drift time values measured from IM-MS data acquired using a drift-tube can be linearly correlated to CCS values36. For travelling wave IM-MS (TWIMS) measurements, calculating CCS values requires a calibration curve obtained from protein standards with known CCS values37.Compact structures travel faster than extended or elongated structures due to reduced interactions with buffer gas in the mobility cell38. Therefore, IM-MS can be used to detect if the native folded structure has been retained in the gas phase39,40. This section outlines how to measure IM-MS and calculate the CCS of protein using TWIMS.
- After optimizing instrument conditions for stable transmission (Step 2), reduce the collisional energy and sampling cone as low as possible whilst retaining good spectra quality.
- Use the optimized wave velocity and wave height to acquire IM-MS (Step 2).
- Measure the ion drift time with IM-MS at three different wave velocities (e.g., 550, 600 and 650 m/s) whilst maintaining the same wave height (e.g., 40 V).
- To determine the protein ions CCS, measure protein calibrants under the same instrument conditions used for protein under investigation. Optimal drift-time calibration requires measurement of proteins with known CCS.
- Select four calibrants, two with a mass above and two with a mass below that of the protein under investigation37. Most importantly, make sure that the wave height and wave velocity are the same as those recorded for the protein under investigation.
- Calculate CCS manually41 or using a specialized software such as PULSAR33 and Amphitrite34 (Figure 5).
- To check whether the protein is native-like in the gas-phase, compare experimental CCS to theoretical CCS obtained from high resolution structures. For HerA-NurA, calculate theoretical CCS using the Projection Approximation (PA) method used in MOBCAL42. Other methods include trajectory method (TM)42 and exact hard sphere scattering (EHS)43.
4. In-Solution Disruption of Protein Complexes for Native MS and IM-MS Led Structure Determination
Note: Protein sub-complexes can in some cases be identified from the same solution as the intact complex. However, further structural information such as inter-subunit connectivity and complex assembly can be attained from disrupting protein interactions in solution, to form sub-complexes. This can be achieved in several ways such as the addition of organic solvent, increasing the ionic strength or manipulating the pH. To gain insight into the HerA-NurA complex subunit connectivity and complex assembly, sub-complexes were generated in solution by adding solvents which perturb subunit interactions.
- Prepare the protein sample and buffer exchange into ammonium acetate as described in Step 1.
- Add 10-40% of solvent in 10% increments. Solvents typically used are Methanol (MeOH), dimethyl sulfoxide (DMSO) or acetonitrile (ACN).
NOTE: This can be performed within a polypropylene microcentrifuge tube. - Incubate the mixture on ice for 1 h.
- Acquire an IM-MS spectrum for each condition (Steps 2 and 3) (Figure 4).
- Use the SUMMIT software44 to assign protein sub-complexes and generate protein interaction networks. Alternatively, manually generate a list of theoretical masses of the expected species.
- To ensure subcomplexes are folded, calculate the experimental CCS values for the sub-complexes and compare to theoretical CCS as explained in Step 3 (Table 1, Figure 5 and Figure 6).
5. Investigating Protein Complex Stability using Collision Induced Unfolding (CIU)
Note: CIU can be used probe the structural stability of proteins and their complexes upon ligand binding. Specialist software packages such as PULSAR33, Amphitrite34 and CIU suite9 can then be used to model the gas-phase unfolding of the protein under investigation with and without ligand. As an example, this section outlines procedure for monitoring gas-phase unfolding trajectories and investigating the stabilizing effect of DNA and ATP binding on the HerA-NurA complex.
- Record IM-MS data whilst increasing the trap acceleration voltage from 10 V to 200 V in 2-10 V increments to progressively unfold the protein in the gas-phase.
NOTE: Recording smaller increments results in more data files to process, however this approach provides more resolved unfolding plot, which is important for analyzing the transition points between folded/unfolded species. - Analyze the data acquired using PULSAR33, Amphitrite34 or CIU suite9 and generate two-dimensional unfolding plots in units of CCS as a function of accelerating voltage (Step 3). For each charge state, this is created by stacking the intensity- normalized CCS distributions at each accelerating voltage (Figure 7 A-Bi).
- Generate a theoretical unfolding plot using one of the software packages. The data will be fitted to an unfolding model. This makes it possible to quantify the collisional energy at which unfolding transitions occur and determine the stability of proteins with and without bound ligands33. An unfolding transition is when a species transitions from one state (based on their experimental CCS values) to another state with a larger CCS.
- To quantitate the transitions, calculate the transitional midpoint(s) between states using algorithms and software such as PULSAR33. This is commonly reported as CV50, which is the collision (trap) voltage value at which 50% of a specific state is depleted.
- Using the CV50 value, calculate the total internal energy of an ion using the center-of-mass collision energy (KECOM)45. KECOM is defined by the total internal energy available for the unfolding transition of an ion and is calculated from the kinetic energy and masses of the collision partners (protein ion and neutral gas) as described in equation (1)10
KEcom (eV) =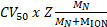 (equation 1).
(equation 1).
Where Z is the ion charge, MN is the mass of the neutral gas and MION is the mass of the protein ion.
NOTE: This is because CIU of proteins is charge dependent46,47. It is recommended to perform the KECOM analysis to more than one charge state ( Figure 7Aii).
6. Modelling Procedures for Differential Molecular Dynamics Simulations used in Integrative MS
NOTE: Using models of protein subunits or complexes such as from crystal structures, differential MD simulations (protein complex with and without ligand) can be used to determine effects of for example ligand presence on protein structure and dynamics. This section details a workflow and tools needed for modelling procedures necessary to set-up differential molecular dynamics simulations.
- Identify the subunits which compose the complex (Figure 8A, in Steps 2 and 3). Source existing models of subunits, e.g., crystal structures from the RCSB databank (https://www.rcsb.org). The UniProt entry of the protein will contain a list of know crystallographic/NMR structures (http://www.uniprot.org). If these are not available, the theoretical sequence can be input to BLAST to identify suitable templates for homology modelling (http://blast.ncbi.nlm.nih.gov/).
- Assemble the complex in the correct topology (Figure 8A-ii). This can be done through various methods. The individual subunits can be fitted into available electron microscopy maps found on the EMDB to assemble the intact complex (https://www.ebi.ac.uk/pdbe/emdb/). A tutorial for fitting PDBs into EM maps using Molecular Dynamics Flexible Fitting (MDFF) can be found here: http://www.ks.uiuc.edu/Training/Tutorials/science/mdff/tutorial_mdff-html/.
- Identify missing regions of the complex (Figure 8A-iii). Perform multiple sequence alignment (MSA) between the PDB and theoretical sequence to identify residues which may be unfitted into crystal structures, or any mutations inherited from crystallographic experiments. MSA can be performed using the webservers such as T-Coffee (http://tcoffee.crg.cat/apps/tcoffee/do:regular).
- Regenerate missing residues via homology modelling (Figure 8A-iv). Missing residues of the protein complex can be built using the MODELLER program (https://salilab.org/modeller/). MODELLER can output an ensemble of n models in different regenerated configurations. Good models can be identified based on their Discrete Optimised Protein Energy (DOPE) score. A comprehensive tutorial is provided on the software website (https://salilab.org/modeller/tutorial/).
- Perform differential molecular dynamics (MD) simulations of the protein complex (Figure 8B) to identify regions of proteins which respond to a particular environmental change, e.g., presence of a ligand. In such simulations, behavioural parameters from Simulation A (protein only) which acts as a reference, is subtracted from Simulation B (protein+ligand). The differential root mean square fluctuation (RMSF) calculated between Simulations A and B can inform on regions of the protein which increase or decrease in flexibility in a ligand dependent manner.
- Perform MD simulations and downstream analysis using GROMACS (http://www.gromacs.org). A tutorial can be found at: http://www.bevanlab.biochem.vt.edu/Pages/Personal/justin/gmx-tutorials/Lysozyme/index.html. To elimate model bias, the structure of the ligand-bound complex should be generated first. The protein is then copied from this without the ligand, to yield a protein model identical to the ligand-bound complex.
Native MS results revealed the oligomeric state, composition and topology of the HerA-NurA complex (Figure 1). As non-covalent interactions are preserved in the gas-phase, native MS of ATP-γ-S and ADP titrations experiments determined the pairwise nucleotide binding to HerA-NurA (Figure 2) and that increasing the ATP-γ-S concentration increases the relative intensity of hexameric HerA (Figure 3). Structural information regarding subunit interactions were obtained from in-solution disruption followed by native MS and were in agreement with and theoretical masses (Figure 4 and Table 1).
The experimental CCS values of proteins and their complexes was derived from IM-MS experiments (Figure 5). These values are rotationally averaged gas-phase cross-sectional calculations of the molecular shape, and describe the dimensional state of the protein. CCS values are compared to theoretical measurements from x-ray crystallography and a good agreement infers that the native shape in retained in the gas-phase (Table 1). This validates using CCS values for building low-resolution models of the protein assembly48.
Experimental CCSs can be calculated for each charge state ion. A native-like protein conformer may give rise to charge state ions with similar CCS values. However, higher charge state ions increased coulombic repulsions which may lead to protein gas-phase unfolding and larger CCS values compared to the theoretical CCSs. The CCS value of the lowest charge state ions are therefore usually used49. For HerA-NurA, in-solution disruption experiments on HerA and HerA-NurA with and without DNA prompted the generation of an assembly pathway starting with monomers then forming the entire hexameric HerA (HerA6)-dimeric NurA (NurA2) complex with DNA (Figure 6).
Differences in the CIU unfolding plots between the apo (ligand-free) and ligand bound define the change in complex stability upon ligand binding. A higher CV50 or KECOM value implies a more stabilized ion in the gas-phase. CIU and KECOM analysis revealed DNA-bound HerA-NurA is more stable than the DNA-free complex (Figure 7Aii). From CIU-MS analysis in the respective ATP-binding states, the four-ATP-γ-S bound state reduced complex stability in the gas-phase and the six -ATP-γ-S bound state where are all sites are occupied was the most stable (Figure 7Bii). Native MS can reveal the discrete nucleotide binding states of HerA; however, it cannot distinguish which HerA subunits are binding ATP and where this binding takes place. This information can be derived from explicit solvent MD simulations on the hexameric HerA and the HerA-NurA following the summarised Workflow (Figure 8).
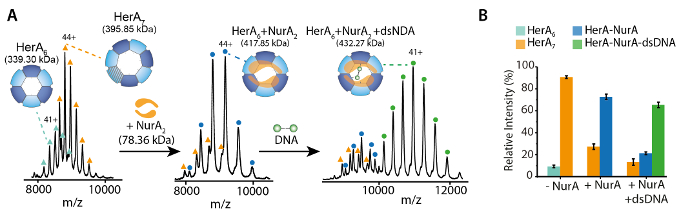
Figure 1. Interrogating the oligomeric state, composition and topology of the HerA-NurA non-covalent complex. (A) Mass spectra of HerA, HerA-NurA and HerA-NurA in the presence of DNA (15.4 kDa 25 bp double- stranded DNA). The HerA sub-complex exists as both a hexamer and a heptamer. NurA dimer binds and to a HerA hexamer imposing oligomeric conversion. The DNA binds the formed HerA- NurA complex (Results adapted from Z. Ahdash et al., 201722). (B) Relative intensities of the identified species are calculated using UniDec32. Please click here to view a larger version of this figure.
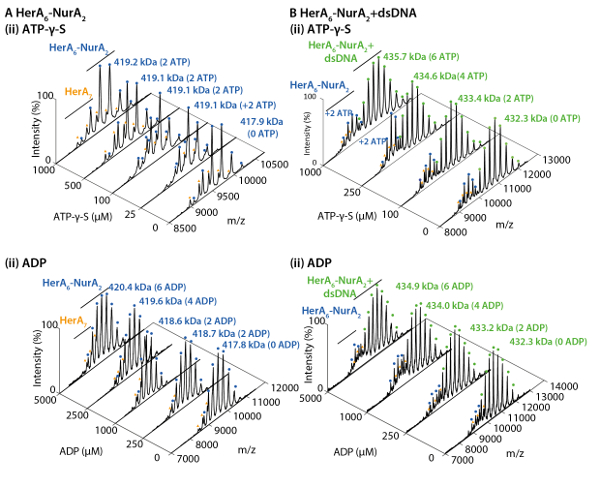
Figure 2. Native ESI-MS reveals the mechanism of nucleotide binding to HerA-NurA. Mass spectra (A) HerA-NurA and (B) HerA-NurA-DNA with increasing concentrations of (i) ATP-γ-S and (ii) ADP. Measured masses are compared to theoretical masses and the amount of ATP-γ-S or ADP bound are determined. Measured masses and number of bound nucleotides are shown on the spectra. ATP-γ-S and ADP titrations experiments determined the pairwise nucleotide binding to HerA-NurA alone and when in complex with DNA indicating a cyclical reaction mechanism (Results adapted from Z. Ahdash et al., 201722). Please click here to view a larger version of this figure.
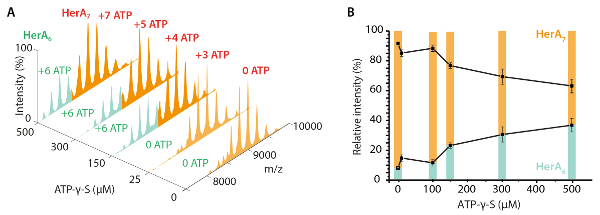
Figure 3. Measuring the effect of increasing ATP-γ-S concentrations on HerA oligomeric state. (A) Mass spectra of HerA at increasing concentrations of ATP-γ-S. (B) Graph showing the relative intensities of different species from native MS calculated using UniDec deconvolution software32. As the ATP-γ-S concentration increases, the relative intensity of hexameric HerA also increases. Number of ATP-γ-S molecules bound is shown on the spectra (Results adapted from Z. Ahdash et al., 201722). Please click here to view a larger version of this figure.
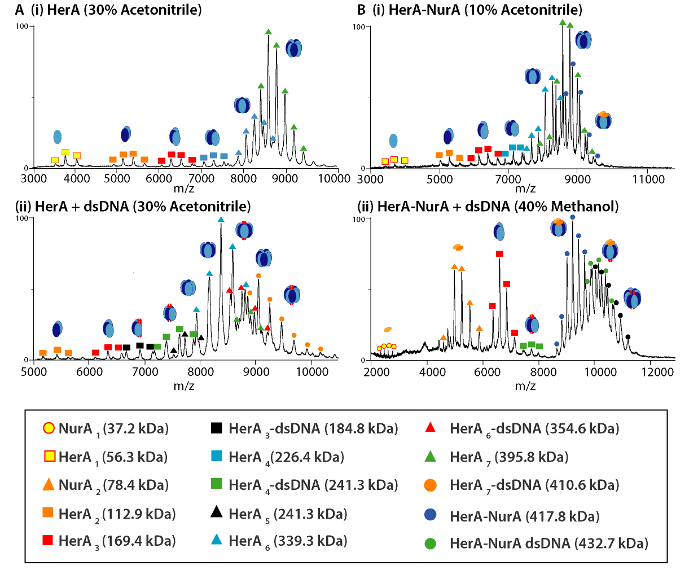
Figure 4. Mass spectra and sub-complex dissociation products of (A) HerA and (B) HerA-NurA (i) alone and in the presence of (ii) DNA following of in-solution disruption. In-solution disruption experiments were performed using 10-40% of Acetonitrile, Methanol (MeOH) or dimethyl sulfoxide (DMSO) and resulted in the formation of various subcomplexes. Please click here to view a larger version of this figure.
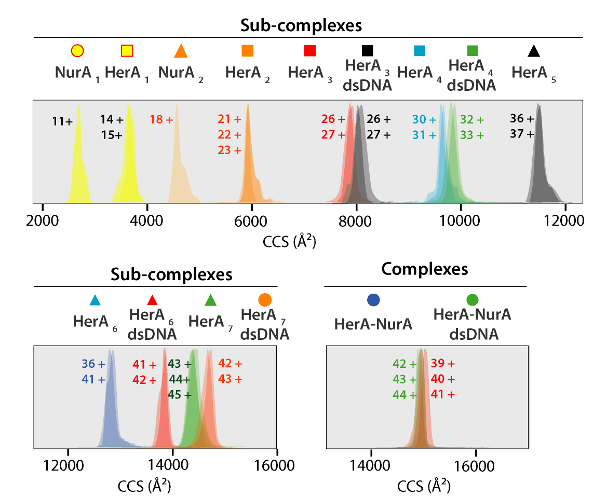
Figure 5. Ion mobility arrival time distributions shown on a CCS axis for complexes and generated sub-complexes. Icon for each sub-complex correlate with those annotated on the spectra in Figure 4. Experimental and calculated masses and CCS values of Sub-complexes are listed in Table 1 all of which showed an agreement between experimental and calculated values (after considering the typical uncertainty in the resolution of travelling wave ion mobility mass spectrometry of ±5-8%37). Please click here to view a larger version of this figure.
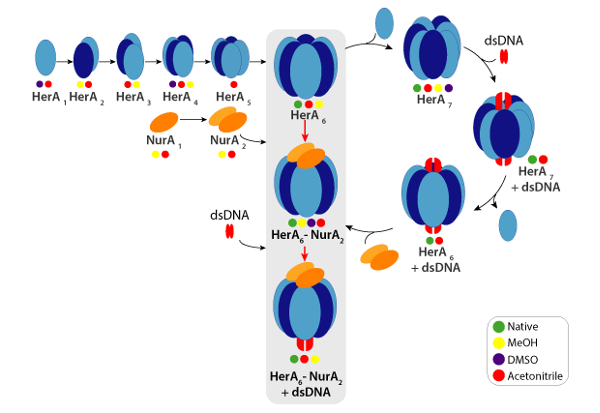
Figure 6. Assembly pathway of the HerA-NurA complex generated from in-solution disruption native MS and IM-MS. Colored circles indicate conditions where each sub-complex was observed: native (green, prior to disruption), Methanol (yellow), DMSO (purple) or Acetonitrile (red). Please click here to view a larger version of this figure.
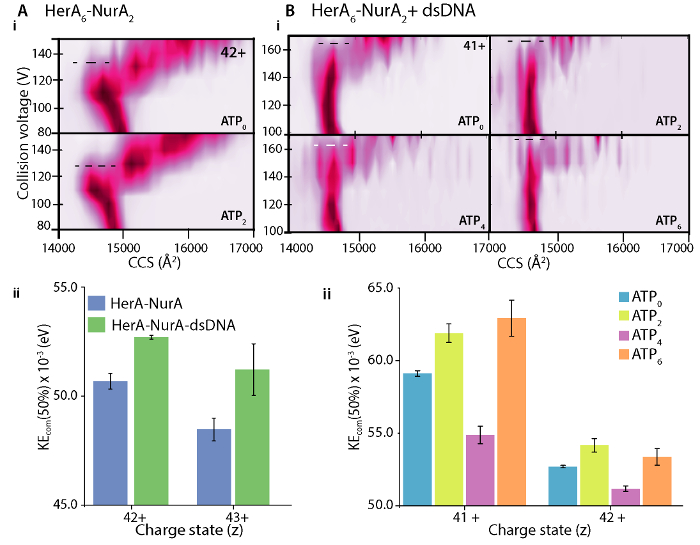
Figure 7. Investigating the stabilizing effect of (A) DNA on HerA-NurA and (B) ATP binding to HerA-NurA-DNA. (i) Gas-phase CIU-MS plots and (ii) center-of-mass collision energies (KEcom) calculation show that that the presence of dsDNA stabilizes the HerA-NurA complex and that the six ATP-γ-S bound state is the most stable. Stabilization for different charge states is shown. Plots were generated using PULSAR 33. Results from (A) adapted from Z. Ahdash et al., 201722. Please click here to view a larger version of this figure.
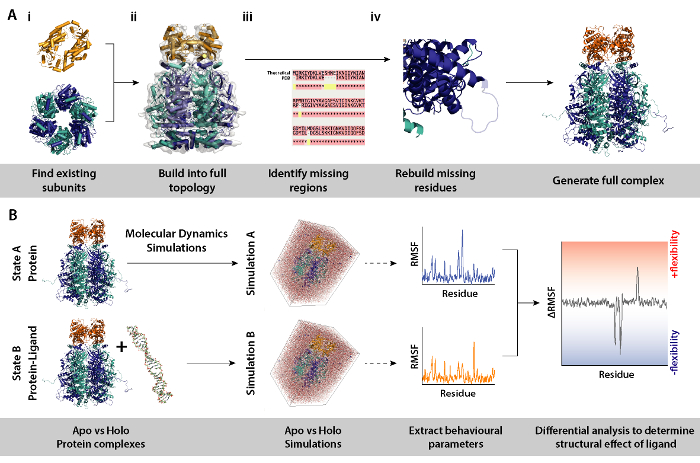
Figure 8. Workflow for the modelling procedures for differential molecular dynamics simulations. (A) Generating the complex under investigation by building the topology from existing subunits and rebuilding missing residues. (B) Workflow for running molecular dynamics simulations on the protein complex with and without ligand. Molecular dynamics simulations are ran for the protein only which acts as a reference and which is subtracted from simulation of protein plus ligand. This is followed by calculating the differential root mean square fluctuation (RMSF) between the simulations and determining the effect of ligand binding. Please click here to view a larger version of this figure.
| Complex / sub-complex | Theoretical Mass (kDa) | Experimental Mass (kDa) | Theoretical CCS (Å2) | Experimental CCS (Å2) [charge] | Condition HN |
| HerA6-NurA2 | 416.22 | 417.85 | 14531 | 14577 [42+] 14599 [43+] 14608 [44+] 14637 [45+] |
10-20% ACN, 10-40% DMSO, 10% MeOH |
| HerA6-NurA2-dsDNA | 431.72 | 432.27 | – | 14661 [39+] 14728 [40+] 14781 [41+] 14837 [42+] |
10% ACN, 10% MeOH |
| NurA1 | 39.12 | 38.18 | 3254 | 2618 [10+] 2746 [11+] 2878 [12+] |
10-40% ACN, 10% MeOH, 20-40% DMSO |
| NurA2 | 78.24 | 78.36 | 4890 | 4903 [16+] 4614 [17+] 4537 [18+] 4666 [19+] |
10-40% MeOH, 20-40% DMSO |
| HerA1 | 56.33 | 56.32 | 4131 | 3647 [14+] 3792 [15+] 3950 [16+] |
10-40% ACN, 40% DMSO |
| HerA2 | 112.66 | 112.95 | 6475 | 5648 [20+] 5747 [21+] 5842 [22+] 5996 [23+] |
40% Meth, 10-40% ACN |
| HerA3 | 168.99 | 169.39 | 8607 | 7501 [25+] 7616 [26+] 7717 [27+] 7867 [28+] |
10-40% MeOH, 10-40% CAN, 40% DMSO |
| HerA3 + DNA | 183.99 | 184.976 | – | 7655 [26+] 7990 [27+] 8107 [28+] |
10-30% ACN |
| HerA4 | 225.32 | 226.2 | 10477 | 9205 [30+] 9287 [31] 9493 [32+] 9961 [33+] |
10-40% MeOH, 10-40% ACN |
| HerA4 +DNA | 240.82 | 241.33 | – | 9637 [31+] 9756 [32+] 9830 [33+] |
10-30% ACN |
| HerA5 | 281.65 | 282.75 | 11853 | 10847 [36+] 10958 [37+] 11161 [38+] |
30-40% ACN |
| HerA6 | 337.98 | 339.3 | 12517 | 12335 [38+] 12386 [39+] 12498 [40+] 12590 [41+] 12676 [42+] 13019 [43+] |
10-40% MeOH, 10-40% ACN |
| HerA6 +DNA | 353.48 | 354.626 | – | 12890 [40+] 13081 [41+] 13184 [42+] 13273 [43+] 13463 [44+] 13576 [45+] |
30% ACN |
| HerA7 | 394.3 | 395.85 | 13901 | 14154 [42+] 14219 [43+] 14261 [44+] 14285 [45+] 14335 [46+] |
10-40% MeOH, 10-40% ACN, 10-40% DMSO |
| HerA7 +DNA | 409.8 | 410.62 | – | 14414 [41+] 14510 [42+] 14558 [43+] 14598 [44+] 14630 [45+] 14641 [46+] |
10% ACN |
Table 1. Experimental and calculated masses and CCS values of HerA-NurA and its sub-complexes generated form in-solution disruption studies.
