Modeling Charcot-Marie-Tooth Disease In Vitro by Transfecting Mouse Primary Motoneurons
Summary
The goal of this technique is to prepare a highly enriched culture of primary motoneurons (MNs) from murine spinal cord. To evaluate the consequences of mutations causing MN diseases, we describe here the isolation of these isolated MNs and their transfection by magnetofection.
Abstract
Neurodegeneration of spinal motoneurons (MNs) is implicated in a large spectrum of neurological disorders including amyotrophic lateral sclerosis, Charcot-Marie-Tooth disease, and spinal muscular atrophy, which are all associated with muscular atrophy. Primary cultures of spinal MNs have been used widely to demonstrate the involvement of specific genes in such diseases and characterize the cellular consequences of their mutations. This protocol models a primary MN culture derived from the seminal work of Henderson and colleagues more than twenty years ago. First, we detail a method of dissecting the anterior horns of the spinal cord from a mouse embryo and isolating the MNs from neighboring cells using a density gradient. Then, we present a new way of efficiently transfecting MNs with expression plasmids using magnetofection. Finally, we illustrate how to fix and immunostain primary MNs. Using neurofilament mutations that cause Charcot-Marie-Tooth disease type 2, this protocol demonstrates a qualitative approach to expressing proteins of interest and studying their involvement in MN growth, maintenance, and survival.
Introduction
Neuromuscular diseases encompass a variety of clinically and genetically distinct pathologies that are characterized by the alteration of muscle and/or the nervous system. Because of advances in sequencing technologies, hundreds of genes responsible for these rare disorders have been identified during the last decade (list available at the Neuromuscular Disease Center, http://neuromuscular.wustl.edu/index.html). The variety of identified mutations indicates that different mutations in a single gene can cause different phenotypes and diseases1,2,3 and that mutations in different genes can produce similar phenotypes4,5. In this context, there are efforts to develop cellular models that can become powerful tools for analyzing mutation consequences and pathological mechanisms.
Spinal MNs have large somas located in the ventral horns of the spinal cord, form long axons to target skeletal muscle fibers, and allow for voluntary movements through the release of acetylcholine at neuromuscular junctions. Since MNs are affected by neuromuscular diseases such as amyotrophic lateral sclerosis, Charcot-Marie-Tooth disease (CMT), and spinal muscular atrophy, Dr. Henderson and colleagues developed the first protocol6 that allowed for cultivation of in vitro spinal MNs and the discovery of neurotrophic factor GDNF7 (glial cell derived neurotrophic factor). Technical refinements since then have allowed for more accurate purification of spinal MNs and their subtypes using FACS8, but enrichment by density gradient remains powerful and widely used in laboratories currently working with primary spinal MNs9,10,11,12,13,14. Subsequently, it is also possible to obtain a higher MN purification grade through immunopanning by taking advantage of surface marker p75(NTR) 15,16,17.
The spinal cord contains different subtypes of cervical, thoracic, and lumbar MNs, as well as median and lateral motor columns that differ among their location in the anterior horn on a dorso-ventral axis and among the targets they innervate8,18. Primary MN cultures can recover all of these MN subtypes in physiological proportions. The main limitation of this technique is the low number of MNs obtained at the end of the procedure; in fact, it can be expected to obtain around 105 MNs from six embryos, which is suitable for microscopy but limiting for biochemistry experiments. To perform experiments with more standardized subtypes and abundant MNs (>106 cells), embryonic stem cell-derived MNs should be considered18.
Transfection of wild-type/mutant transgenes or knockdown endogenous genes into primary MNs is a rapid and helpful tool for deciphering physiopathological pathways, especially when mouse models are unavailable. Magnetofection is one technique for transfecting primary neurons, similar to lipofection without the related neurotoxicity. Furthermore, transfection can be performed on mature neurons after several days in vitro, unlike techniques based on electroporation9. However, one disadvantage of this technique is that the beads bind nucleic acids in the culture, causing noise in DAPI labeling. Viral infection is likely the most efficient technique for transfecting MNs; however, magnetofection does not require certain safety procedures needed for viral production and cellular infection.
Protocol
All procedures involving animals were accepted by the ethical committee of the institution.
1. Solution Preparation
- Prepare 10 mL of 4% BSA dialyzed in L-15 medium.
- Dissolve bovine serum albumin powder at 4% (w/v) in 10 mL of L-15 medium.
- Add the solution to a 20 mL dialysis cassette with a 20 kDa cut-off. Dialyze against 500 mL of L-15 medium for 3 days under agitation at 4 °C. Change the L-15 medium every day.
- Filter the solution through a 0.22 µm membrane and aliquot in 15 mL tubes. Store the tubes at -20 °C.
NOTE: This protocol uses the following references: unmodified raw L-15 medium = L-15 medium, acidic L-15 medium = pH L-15, and basic L-15 medium = Bicarbonate L-15.
- Prepare 200 mL of pH L-15.
- Adjust the pH of the L-15 medium: First, fill a wash-bottle with some pieces of dry ice. Inject gas (CO2) into the L-15 medium until the color turns orange (~pH 6.4 and ~40 pressures). The pH can be also adjusted with a standard pH meter.
- Filter the medium through a 0.22 µm membrane and store at 4 °C for one month.
- Prepare 100 mL of Bicarbonate L-15.
- Add 2.5 mL of 7% sodium bicarbonate to 97.5 mL of L-15 medium and store at 4 °C.
- Prepare the IPCS (Insulin Putrescin Conalbumin Selenite) supplements.
- Dissolve 2.5 mg of insulin in 0.5 mL of 0.1 M HCl. Add 4.5 mL of double-distilled water.
- Dissolve 8 mg of putrescine in 5 mL of phosphate-buffered saline (PBS).
- Dissolve 100 mg of conalbumin in 10 mL of PBS.
- Dissolve 1 mg of sodium selenite in 19.3 mL of water, adjust the pH to 7.4, and dilute the solution 10-fold.
- Combine 1 mL of insulin solution, 1 mL of putrescine solution, 1 mL of conalbumin, and 0.1 mL of sodium selenite solution to obtain 3.1 mL of IPCS supplement solution. Store the solution at -20 °C.
- Prepare a 0.02 mM progesterone solution.
- Dissolve 1 mg of progesterone in 1.6 mL of 80% ethanol.
- Dilute the solution 100-fold in 100% ethanol and store it at -20 °C.
- Prepare 100 mL of complete L-15 medium.
- Add 1.8 mL of D-glucose solution (200 mg/mL) to 92 mL of pH L-15 to obtain a final concentration of 3.5 mg/mL glucose.
- Combine 1 mL of 100x Penicillin-Streptomycin (10,000 U/mL), 2 mL of heat-inactivated horse serum, 3.1 mL of IPCS mix, and 0.1 mL of progesterone solution (2 x 10-5 M).
- Filter the medium on a 0.22 µm membrane and store it at 4 °C.
- Prepare 5 mL of density gradient solution (final concentration of 5.5%) per experiment.
- Add 476 µL of the commercial 60% density gradient medium to 4524 µL of L-15 (if possible, without red phenol to increase the visual contrast at the interphase; dilution ratio of 1:10.5).
- Store the solution at 4 °C for 2 weeks or freeze it at -20 °C.
- Prepare 10 mL of culture media.
- Add 200 µL of supplement medium to 9.6 mL of neuron cell culture medium.
- Add 200 µL of heat-inactivated horse serum (which tends to differentiate glial cell precursors and works against proliferation) to 25 µL of glutamine and 10 µL of 2-mercaptoethanol.
- Filter the media through a 0.22 µm filter.
- Add the following neurotrophic factors: ciliary neurotrophic factor (CNTF) at a final concentration of 10 ng/mL, and brain derived neurotrophic factor (BDNF) and glial cell derived neurotrophic factor (GDNF) at final concentrations of 1 ng/mL.
- Store the media at 4 °C.
- Prepare 50 mL of dissection media.
- Add 2.25 mL of glucose (200 mg/mL) to 47.05 mL of HBSS. Add 700 µL of 1 M HEPES with pH 7.4. Store the media at 4 °C.
- Prepare a 4% PFA solution.
- Dissolve 20 g of PFA in 500 mL of PBS.
- Under a chemical hood, heat the solution to 60 °C and add a few drops of 1 M NaOH until the solution becomes clear.
- Filter the solution through a filter paper and adjust the pH to 7.2. Store it at -20 °C.
NOTE: Alternatively, other commercial PFA solutions can be used and diluted to 4% in PBS.
- Clean the dissection tools including the forceps, scissors, and silicone dishes with 70% ethanol before use, and with soap and clear water after use.
2. Poly-ornithine/Laminin (Po/L) Dish Coating
NOTE: Volumes are adapted to coat a 24-well plate.
- If needed, put a sterile 12 mm coverslip in the well.
- Coat the wells with 500 µL of 10 µg/mL Poly-L-Ornithine solution dissolved in water.
- Let the wells settle at room temperature for 1 h.
- Remove the Poly-L-Ornithine solution and air-dry for 30 min.
- Coat the wells overnight at 37 °C with 500 µL of 3 µg/mL laminin in bicarbonate L-15.
NOTE: Wells can be stored at 37 °C for 7 days in the incubator. For long incubations, adding sterile PBS between the wells can help to avoid medium evaporation.
3. Dissection
- Sacrifice a pregnant mouse when the embryos are at embryonic stage E12.5. Clean the belly with 70% ethanol.
- Remove the womb by cutting the two oviducts and the vagina and put it in a Petri dish filled with cold PBS (without Ca2+ Mg2+).
- Collect all the embryos and transfer them to fresh, cold PBS (without Ca2+ Mg2+) in a Petri dish.
- Put one embryo in a dish coated with silicone and full of dissection media (with HBSS, 4.5 g/L glucose, and 7 mM HEPES pH 7.4).
NOTE: Embryo dissection is performed under a microscope using clean fine forceps and scissors. Alternatively, a regular Petri dish without silicone can be used for dissection. - Remove the head, tail, and viscera, using forceps as scissors.
- Maintain the embryo with the belly against the silicone with forceps.
- With a second pair of forceps, insert one of the tips into the central canal of the rostral spinal cord.
- Close the forceps to tear the dorsal tissue a few millimeters.
- Repeat this operation toward the caudal side until the entire spinal cord is open (Figure 1B).
- Detach the rostral part of the spinal cord (cerebral trunk) from the body.
- Turn the embryo to maintain the open spinal cord against the silicone.
- Pinch the rostral part of the spinal cord and pull the embryo by the head to extract the spinal cord.
NOTE: Meninges and dorsal root ganglia (DRGs) should remain attached to the embryo. Otherwise, carefully pull away any remaining meninges and DRGs still attached to the spinal cord. At this step, DRGs could also be collected for sensitive neuron culture (see Discussion). - Flatten the spinal cord on the silicone and remove the dorsal half of the cord by cutting along the middle of each side using a scalpel. Cut the middle part into 10 pieces using a scalpel, and transfer the pieces to a new tube.
4. Spinal Cord Cell Suspension
- Repeat this dissection procedure for 6 to 8 spinal cords.
- Let the spinal cord pieces collect at the bottom of the tube and replace the HBSS with 1 mL of Ham-F10 medium.
- Add 10 µL of trypsin (2.5% w/v; final concentration 0.025%). Incubate the tube for 10 min at 37 °C.
- To stop the enzymatic digestion, transfer the fragments to a new 15 mL tube containing 800 µL of complete L-15 medium, 100 µL of 4% (w/v) BSA, and 100 µL of DNase (1 mg/mL in L-15 medium).
- Triturate twice by aspirating 1 mL using a P1000 pipette. Let the fragments settle for 2 min. Collect the supernatant in a fresh 15 mL tube.
- To the fragments, add 900 µL of complete L-15 medium, 100 µL of 4% (w/v) BSA, and 100 µL of DNase (1 mg/mL in L-15 medium).
- Triturate 6 times by aspirating 1 mL using a P1000 pipette. Let the fragments settle for 2 min. Add the supernatant to the 15 mL tube obtained in step 4.5.
- To the fragments, add 900 µL of complete L-15 medium, 100 µL of 4% (w/v) BSA, and 100 µL of DNase (1 mg/mL in L-15 medium).
- Triturate 10 times by aspirating 1 mL using a P1000 pipette. Let the fragments settle for 2 min. Add the supernatant to the 15 mL tube obtained in step 4.5. Proceed with the supernatant.
- Using a P1000 pipet, gently place a 2 mL BSA 4% (w/v) cushion at the bottom of supernatant tube. Centrifuge at 470 x g for 5 min.
- Remove the supernatant and resuspend the cell pellet with 2 mL of complete L-15 medium.
5. MN Enrichment by Gradient Density
- Split the cell suspension into two 15 mL tubes (1 mL each). Then, add 2 mL of complete L-15 medium. If starting with 6 embryos, use the equivalent of 3 embryos per tube in 3 mL of medium.
- Add 2 mL of density gradient solution by slowly adding the solution to the bottom of each tube to obtain a sharp interface.
- Centrifuge at 830 x g for 15 min at room temperature. After this step, small cells should be at the bottom of the tube, whereas large cells such as MNs should be at the density gradient solution/medium interface.
- Collect the cells at the interface by aspirating 1 or 2 mL and transfer them to a fresh 15 mL tube. Adjust the volume to 10 mL with L-15 pH medium.
- Add 2 mL of the 4% BSA cushion to the bottoms of the tube and centrifuge at 470 x g for 5 min at room temperature.
- Remove the supernatant and resuspend the pellet in 3 mL of complete L-15 medium.
- Repeat step 5.5.
- Remove the supernatant and resuspend the cell pellet in 2 mL of complete culture medium. Count the cell number and viability under a phase contrast microscope with a hemocytometer.
NOTE: For 6 spinal cords, the cell number is expected to be around 1 x 105 MNs.
6. MN Culture
- Dilute the MNs to the appropriate density, usually 5,000 to 10,000 cells in 500 µL of culture medium per well in a 24-well plate.
NOTE: Seeding density is around 30,000 and 1,500 cells for 6- and 96-well plates, respectively. - Remove the laminin solution with a P1000.
- Immediately transfer the MNs diluted in culture medium into the coating plates to avoid drying.
- Incubate the culture for 2 days at 37 °C.
7. Magnetofection of MNs
NOTE: In the following steps, the quantities used are meant for the transfection of one well of a 24-well plate. Please refer to the manufacturer's protocol for another format.
- For the DNA preparation, resuspend 1 µg of DNA in 50 µL of neuronal culture medium and vortex for 5 s.
- For bead tube preparation, resuspend 1.5 µL of beads in 50 µL of neuronal culture medium.
- Add the 50 µL bead solution to the 50 µL DNA solution and incubate for 20 min at room temperature (total volume = 100 µL).
- During this incubation, withdraw 100 µL of culture medium from the well that will be transfected.
NOTE: Put the magnetic plate in the incubator to warm it to 37 °C. - Transfer 100 µL of the DNA/bead mix to the well, and incubate the plate 20–30 min on the magnetic plate at 37 °C.
- Wait at least 24 h before detection of cDNA expression.
8. Fixation and Staining
- Wash the culture 2 times with PBS.
- Incubate the culture for 20 min at room temperature in 4% PFA/PBS.
- Wash the culture 2 times with PBS.
- Incubate the culture in 500 µL of blocking buffer (PBS, 4% BSA, 2% normal serum, 0.3% Triton X-100, 0.1 M glycine) for 1 h at room temperature.
NOTE: Normal serum should be derived from the same species as the secondary antibody (e.g., Normal Goat Serum). - Incubate the culture overnight at 4 °C with primary antibody diluted in 250 µL of blocking buffer.
NOTE: For example, TUJ1 is used at 1/1000, SMI32 is used at 1/1,000, and Lc3b is used at 1/200. - Wash the culture 3 times with PBS.
- Incubate the culture with fluorophore-conjugated secondary antibodies diluted in 250 µL of blocking buffer for 1 hour at room temperature.
- Wash the culture 3 times with PBS.
- Mount on glass slides using mounting medium.
Representative Results
After 24 hours in the culture, motoneurons (MNs) should already show significant axonal growth (at least 6 times longer than the soma size). In the following days, axons should continue to grow and display branching (Figure 2). There will be different morphologies due to subtype specificities. For example, median column MNs that innervate axial muscles have shorter and more branched axons than lateral motor column MNs that innervate limb muscles18.
These isolation and culturing techniques described above have recently been used to characterize a new mutation in the neurofilament gene NEFH that causes an autosomal dominant axonal form of CMT13. In this case, two days after plating, purified MNs were transfected with plasmids encoding either a mutant form of NEFH fused to eGFP or the wild-type form fused to eGFP. Two days later, mutant NEFH-eGFP formed aggregates along the cytoskeletal network. Four days after magnetofection, these aggregates evolved in a prominent perinuclear aggresome containing the LC3b autophagic marker (Figure 3). This approach allowed us to demonstrate that the mutant form of NEFH induced the formation of aggregates that are associated with autophagic pathways in primary MNs.
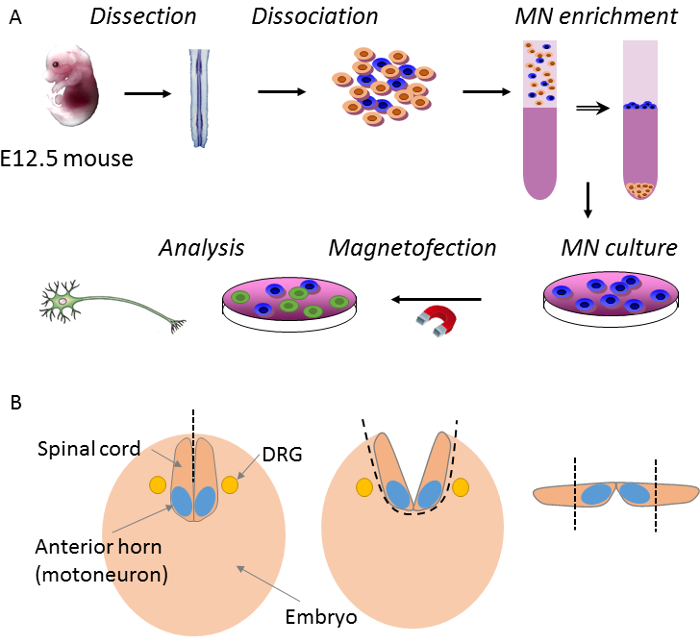
Figure 1: Overview of procedure main steps. (A) E12.5 mouse embryos are dissected to extract the ventral horn of the spinal cord. Then, neurons from the ventral horn cells are dissociated and MNs are purified by density gradient before plating. Finally, MNs are transfected by magnetofection and analyzed after a few days. (B) The first step in spinal cord dissection is when the roof plate of the spinal cord is opened along the rostro-caudal axis with forceps. Then, the spinal cord is detached from the body by pulling the cerebral trunk. At this point, meninges and dorsal root ganglia (DRG) remain on the embryonic body. The dorsal part of the spinal cord is cut longitudinally with a scalpel. Please click here to view a larger version of this figure.
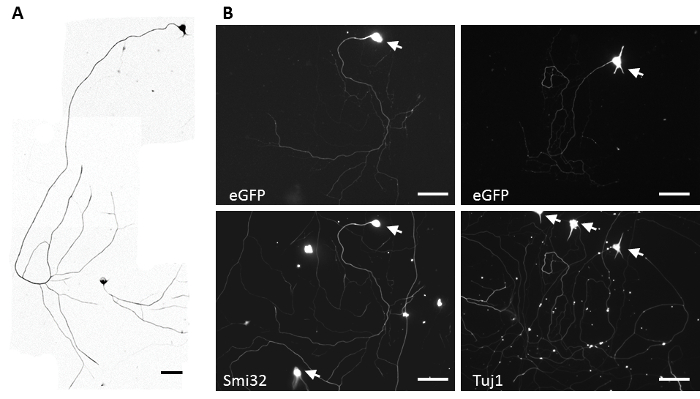
Figure 2: Representative MN culture. (A) MNs morphology after 4 days of culture revealed by staining of endogenous non-phosphorylated neurofilament heavy chain (SMI32). Scale bar = 100 µm. (B) MNs morphology after 4 days in vitro and transfected for eGFP transgene by magnetofection at day 2. MNs were counterstained with the marker SMI32 or the pan-neuronal marker Tuj1. Arrows show somas of the neurons. Scale bar = 100 µm. Please click here to view a larger version of this figure.
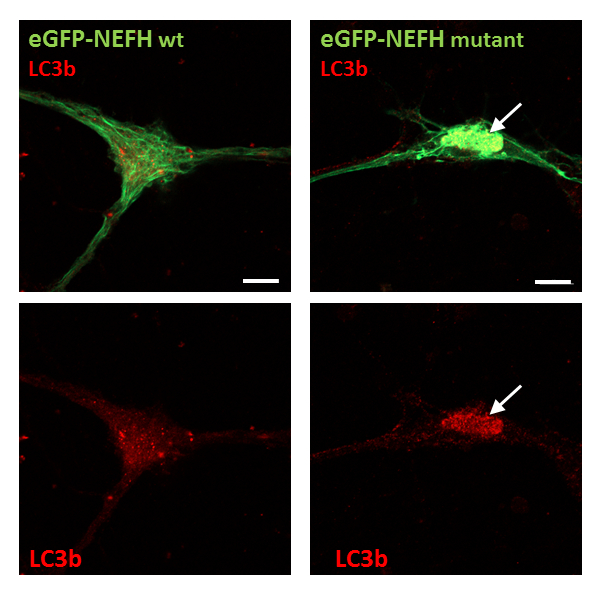
Figure 3: Confocal images of magnetofected MNs expressing wild-type or mutant eGFP-NEFH constructs (in green) and stained for the autophagic marker LC3b (in red). Arrows show mutant eGFP-NEFH protein aggregates that are also LC3b positive. Scale bar = 10 µm. Images are modified from Jacquier et al. 2017, Acta Neuropathologica Communication13.
Discussion
One of the critical points in this protocol is that the mouse embryos are dissected at a precise time window during development (E12.5) to optimize the amount of MNs obtained at the end. In addition, for optimal yield, the dissection should be performed in the morning or early in the afternoon. Before E12.5 (e.g., at E11.5), dissection is difficult, especially regarding the elimination of the meninges. After E12.5, the number of obtained MNs drops significantly. To control the embryo stage of development, adult females and males are first placed together at the end of the day. The next morning, the adults are separated, and females are checked for the presence of a vaginal plug. If a plug is present, embryos are at this point considered to be in E0.5 day of development. For these experiments, we typically use a mouse line that is vigorous and productive. Progenitors originating from a colony bred in Missouri were used in this protocol, and the strain was named CF1 (Carworth Farms strain 1). This strain was introduced at Charles River Laboratories France in 1967, and it acquired the name OF1 (Oncins France 1).
A second critical point is the centrifugation steps that greatly influence the purity of the culture. MNs represent a low percentage of the spinal cord cells (around 1%), and the goal is to obtain a culture containing 40 to 50% MNs among other spinal neuronal cells and less than 5% glial cell progenitors. To monitor the efficacy of centrifugations, cell suspension samples acquired before and after each step can be plated in MNs medium for 24 hours, followed by fixing and staining for the expression of motor neuron markers Hb9 (81.5C10, DSHB) or Islet-1/Islet-2 (ISL1/2, 40.2D6 /39.4D5, DSHB), whose combinations define different MNs subpopulation19. On more mature MNs, other markers such as Choline Acetyl Transferase (CHAT, Ab144P) or Neurofilament H non-phosphorylated (SMI-32P) can be used to estimate the quality of the culture. A good alternative for quickly monitoring the efficacy of purification is to use transgenic mice that express the Green Fluorescent Protein (GFP) specifically in MNs. For example, Wichterle and colleagues previously developed transgenic mice expressing GFP under the control of the mouse Hb9 (also called Hlxb9 or Mnx1) promoter18. Hb9::GFP transgenic mice display distinct expression of GFP in dendrites, axons, and somas of spinal MNs from embryonic day 9.5 to postnatal day 10.
MNs require growth on a permissive substrate obtained by Poly-L-Ornithine and laminin coating. Depending on experimental conditions, a variety of plastic-, polymer-, and glass-based containers can be used (e.g., 6-, 24-, 96-well plates, glass coverslips, or chamber slides). Among these options, microfluidic devices may be of interest to address specific questions regarding synapse formation or axonal guidance and transport20. MNs are known to be sensitive to changes in their microenvironment involving patterns, concentration factors, mechanical changes in the substrate (e.g., stiffness), sheer stress, and spatiotemporal gradient cues (e.g., topographic features, patterning of surfaces with substances of different cellular affinities). Microfluidic platforms are systems that can integrate multifactorial conditions, allowing for the determination of optimal cellular microenvironments.
Since the discovery of GDNF's survival effects7, the number of neurotrophic factors involved in MN initial growth and survival, guidance and development of axons, and inducing of synaptic plasticity is growing. Interestingly, each factor acts on a specific subpopulation of MNs8. For example, CNTF has been described to protect the median motor column (MMC) which innervates axial muscles, whereas HGF acts on the lateral motor column (LMC) which innervates limb muscles. On the other hand, GDNF and BDNF both show the strongest pro-survival activity on whole populations of MNs. Finally, the combination of GDNF, BDNF, and CNTF is sufficient for protecting more than 70% of a MN population and is commonly used in laboratories.
In standard culture conditions, MNs can be kept in vitro up to 14 days by replacing half of the culture media with fresh media once every 3 days, starting from the fifth day of culture. During in vitro maturation, MNs can be transfected by magnetofection at any time using this protocol. Efficiency and toxicity of this technique may be modulated by the nature of the plasmids and inserts. In our case, when magnetofection was performed on MN culture after 2 days in vitro using a 9 kb plasmid with a CAGGS promotor (pCAGEN21) controlling neurofilament cDNA, we observed an average of 31.48% ± 9.94 (standard deviation) of transfected MNs 48 hours post-transfection. Nevertheless, for other constructs, a different ratio of DNA/beads should be tested as described in the manufacturer's protocol. Another parameter that may influence the transfection level efficiency is the maturation of MNs over time. Indeed, during the first 3 days of culture, growth of axons and enlargement of somas will occur, which increases the surface area where the beads will penetrate. This parameter may increase MN transfection efficiency over time during maturation.
Finally, in comparison with MN cultures, it would be interesting to cultivate sensory neurons22,23,24. In particular, Charcot-Marie-Tooth disease is a sensory motor neuropathy characterized by distal muscular atrophy related to the degeneration of both spinal MNs and sensory neurons of DRGs. Interestingly, DRGs are located on both sides of the spinal cord and can be collected during the same dissection procedure used to obtain MNs. In this regard, it is possible to compare the pathophysiological mechanisms at work in these two relevant cell types, motoneurons and DRG sensitive neurons.
Disclosures
The authors have nothing to disclose.
Acknowledgements
We would like to thank the "Association pour le développement de la neurogénétique" for Dr. Jacquier's fellowship and AFM-Telethon for its support through MyoNeurAlp strategic plan. We would also like to thank Dr. Chris Henderson, Dr. William Camu, Dr. Brigitte Pettmann, Dr. Cedric Raoul, and Dr. Georg Haase, who participated in developing and improving the technique and spread their knowledge.
Materials
| Material | |||
| Silicone dissection dish | Living systems instrumentation | DD-90-S-BLK-3PK | Sylgard |
| round coverslip | NeuVitro, Knittel glass | GG-12-Pre | 12 mm |
| Slide a Lyzer cassettes | ThermoFisher Scientific | 66030 | 20,000 MWCO ; 30 mL |
| Filter unit | Millipore | SCGVU02RE | |
| GP Sterile Syringe Filters | Millipore | SLGP033RS | |
| 4 well plate | ThermoFisher Scientific | 167063 | Nunclon Delta treated plate |
| forceps | FST by Dumont | 11252-20 | #5 forceps |
| scissor | FST by Dumont | 14060-10 | fine scissors |
| scalpel | FST by Dumont | 10035-20 | curved blade |
| scalpel | FST by Dumont | 10316-14 | micro-knife scalpel |
| Petri dish | Greiner | 663102 | ø x h = 100 x 15 mm |
| 15 mL polypropylene tube | Falcon | 352096 | |
| filter paper | Watman | 1001125 | circle, 125 mm diameter |
| glass chamber slide | Lab-Tek | 154526 | 4 chambers |
| Plasmid pCAGEN | Addgene | #11160 | |
| Name | Company | Catalog Number | Comments |
| Solutions and mediums | |||
| Bovine serum albumin | Sigma-Aldrich | A9418 | |
| L-15 medium | ThermoFisher Scientific | 11415056 | |
| L-15 medium, no red phenol | ThermoFisher Scientific | 21083027 | |
| Insulin | Sigma-Aldrich | I6634 | |
| Putrescine | Sigma-Aldrich | P5780 | |
| Conalbumin | Sigma-Aldrich | C7786 | |
| Sodium selenite | Sigma-Aldrich | S5261 | |
| Progesterone | Sigma-Aldrich | P8783 | |
| Poly-DL-Ornithine | Sigma-Aldrich | P8638 | |
| Laminin | Sigma-Aldrich | L2020 | |
| trypsin 2.5%, 10x | ThermoFisher Scientific | 15090046 | |
| DNAse | Sigma-Aldrich | DN25 | |
| PBS w/o Ca Mg | ThermoFisher Scientific | 14190144 | without Mg2+ Ca2+ |
| sodium bicarbonate | ThermoFisher Scientific | 25080094 | |
| Neuron cell culture medium | ThermoFisher Scientific | A3582901 | Neurobasal Plus medium |
| HBSS | Sigma-Aldrich | H6648-500ML | |
| HEPES buffer 1 M | ThermoFisher Scientific | 15630056 | |
| Density gradiant medium | Sigma-Aldrich | D1556 | Optiprep |
| supplement medium | ThermoFisher Scientific | A3582801 | B-27 Plus |
| Horse serum heat inactivated | ThermoFisher Scientific | 26050-088 | |
| L-Glutamine 200 mM | ThermoFisher Scientific | 25030024 | |
| 2-mercaptoethanol | ThermoFisher Scientific | 31350010 | |
| penicilline/streptomycine | ThermoFisher Scientific | 15140122 | 10,000 U/ml |
| Name | Company | Catalog Number | Comments |
| Immuno fluorescence | |||
| PBS, 10x | ThermoFisher Scientific | X0515 | without Mg2+ Ca2+ |
| Paraformaldehyde (PFA) | Sigma-Aldrich | 441244 | |
| normal goat serum | Sigma-Aldrich | G6767 | |
| glycine | Sigma-Aldrich | G7126 | |
| Triton X-100 | Sigma-Aldrich | T8787 | |
| Choline Acetyl Transferase (CHAT) | Chemicon | Ab144P | |
| Neurofilament H non phosphorylated (SMI32) | Biolegends | SMI-32P | IF at 1/1000 |
| Islet-1 | DSHB | 40.2D6 | |
| Islet-2 | DSHB | 39.4D5 | |
| Hb9 | DSHB | 81.5C10 | |
| Vectashield mounting medium | Vector Lab | H-1000 | |
| Beta3 tubulin (Tuj1 clone) | Biolegends | 801201 | IF at 1/1000 |
| Lc3b | Cell Signaling Technology | #2775 | IF at 1/200 |
References
- Gonzalez, M. A., et al. A novel mutation in VCP causes Charcot-Marie-Tooth Type 2 disease. Brain. 137 (11), 2897-2902 (2014).
- Johnson, J. O., et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron. 68 (5), 857-864 (2010).
- Watts, G. D. J., et al. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nature Genetics. 36 (4), 377-381 (2004).
- Brown, R. H., Al-Chalabi, A. Amyotrophic Lateral Sclerosis. New England Journal of Medicine. 377 (2), 162-172 (2017).
- Taylor, J. P., Brown, R. H., Cleveland, D. W. Decoding ALS: from genes to mechanism. Nature. 539 (7628), 197-206 (2016).
- Henderson, C. E., Bloch-Gallego, E., Camu, W. Purified embryonic motoneurons. Nerve Cell Culture: A Practical Approach. , 69-81 (1995).
- Henderson, C. E., et al. GDNF: a potent survival factor for motoneurons present in peripheral nerve and muscle. Science. 266 (5187), 1062-1064 (1994).
- Schaller, S., et al. Novel combinatorial screening identifies neurotrophic factors for selective classes of motor neurons. Proceedings of the National Academy of Sciences. 114 (12), E2486-E2493 (2017).
- Jacquier, A., et al. Alsin/Rac1 signaling controls survival and growth of spinal motoneurons. Annals of Neurology. 60 (1), 105-117 (2006).
- Raoul, C., et al. Chronic activation in presymptomatic amyotrophic lateral sclerosis (ALS) mice of a feedback loop involving Fas, Daxx, and FasL. Proceedings of the National Academy of Sciences. 103 (15), 6007-6012 (2006).
- Madji Hounoum, B., et al. Wildtype motoneurons, ALS-Linked SOD1 mutation and glutamate profoundly modify astrocyte metabolism and lactate shuttling. Glia. 65 (4), 592-605 (2017).
- Magrane, J., Sahawneh, M. A., Przedborski, S., Estevez, A. G., Manfredi, G. Mitochondrial Dynamics and Bioenergetic Dysfunction Is Associated with Synaptic Alterations in Mutant SOD1 Motor Neurons. Journal of Neuroscience. 32 (1), 229-242 (2012).
- Jacquier, A., et al. Cryptic amyloidogenic elements in mutant NEFH causing Charcot-Marie-Tooth 2 trigger aggresome formation and neuronal death. Acta Neuropathologica Communications. 5, (2017).
- Aebischer, J., et al. IFNγ triggers a LIGHT-dependent selective death of motoneurons contributing to the non-cell-autonomous effects of mutant SOD1. Cell Death and Differentiation. 18 (5), 754-768 (2011).
- Wiese, S., et al. Isolation and enrichment of embryonic mouse motoneurons from the lumbar spinal cord of individual mouse embryos. Nature Protocols. 5 (1), 31-38 (2010).
- Camu, W., Henderson, C. E. Purification of embryonic rat motoneurons by panning on a monoclonal antibody to the low-affinity NGF receptor. Journal of Neuroscience Methods. 44 (1), 59-70 (1992).
- Conrad, R., Jablonka, S., Sczepan, T., Sendtner, M., Wiese, S., Klausmeyer, A. Lectin-based Isolation and Culture of Mouse Embryonic Motoneurons. Journal of Visualized Experiments. (55), (2011).
- Wichterle, H., Lieberam, I., Porter, J. A., Jessell, T. M. Directed differentiation of embryonic stem cells into motor neurons. Cell. 110 (3), 385-397 (2002).
- Francius, C., Clotman, F. Generating spinal motor neuron diversity: a long quest for neuronal identity. Cellular and Molecular Life Sciences. 71 (5), 813-829 (2014).
- Neto, E., et al. Compartmentalized Microfluidic Platforms: The Unrivaled Breakthrough of In Vitro Tools for Neurobiological Research. The Journal of Neuroscience. 36 (46), 11573-11584 (2016).
- Matsuda, T., Cepko, C. L. Electroporation and RNA interference in the rodent retina in vivo and in vitro. Proceedings of the National Academy of Sciences of the United States of America. 101 (1), 16-22 (2004).
- Sleigh, J. N., Weir, G. A., Schiavo, G. A simple, step-by-step dissection protocol for the rapid isolation of mouse dorsal root ganglia. BMC Research Notes. 9, (2016).
- Yu, L., et al. Highly efficient method for gene delivery into mouse dorsal root ganglia neurons. Frontiers in Molecular Neuroscience. 8 (2), (2015).
- Malin, S. A., Davis, B. M., Molliver, D. C. Production of dissociated sensory neuron cultures and considerations for their use in studying neuronal function and plasticity. Nature Protocols. 2 (1), 152-160 (2007).
