1. Primer Design and Analysis for T7 Phage Genomic DNA
- Design primers for amplification of T7 phage genomic DNA.
NOTE: F (forward) and R (reverse) primers (see Table of Materials) amplify the T7 DNA sequence located upstream of the library variable region (Figure 1). - Choose an appropriate primer analyzer to evaluate parameters of the primers, including melting temperature (Tm), percent GC content (GC%), primer dimers, hairpin formation and PCR suitability (Figure 1).
- Order the primer oligonucleotides (oligos), and upon arrival, resuspend the lyophilized oligos in DNase and RNase free ultra-pure water to make 100 µM stock concentrations in a 1.5 mL centrifuge tube, and store at -20 °C for several months.
- Make several aliquots (around 50 µL in each 1.5 mL tube) of the 100 µM primer stock.
NOTE: This step is used to prevent frequent freeze-thaw cycles.
2. Prepare Standards for Quantification of T7 Phage Genomic DNA
- Serially dilute 0.1 µg/µL (1.0 x 108 fg/µL) reference T7 phage DNA (purchased; at a known concentration of 0.1 µg/µL, see materials) 10-fold from 1.0 x 106 to 1.0 x 100 fg/µL with ultra-pure H2O in 0.2 mL PCR tubes. Prepare each concentration in a total volume of 20 µL (Figure 2).
- Vortex the PCR tubes to ensure thorough mixing at each step of dilution. Also, change tips when adding the DNA sample to the following dilution.
NOTE: It is recommended to prepare new solution of DNA standards for a standard curve for every batch of the qPCR experiment. - Use the equation 1 (below) to convert for T7 phage reference DNA concentrations from fg/µL to genome copies (gc) /µL (Figure 2).
- T7 phage DNA concentration in gc/µL=
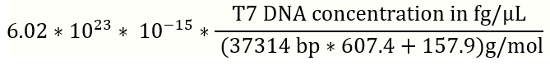 (1)
(1)
NOTE: bp: base pair; reference T7 genomic DNA is 37314 bp.
3. Pre-treatment of Phage Samples Before qPCR Reaction Preparation
- (Option 1) Pipet 20 µL of one T7 clone (phage) selected from biopanning against in vitro cystic fibrosis (CF)-like mucus model into 1.5 mL centrifuge tubes. Add 1.25 units of DNase I (0.5 µL) to each 20 µL sample.
NOTE: T7 phage cysteine constrained heptapeptide library (CX7C) was biopanned against CF-like mucus coated on 24-well membrane inserts (see Table of Materials) for 15 min. Further details are provided in the results section. One T7 clone was chosen for qPCR sample preparation from the eluate.- Option 2: Pipet 200 µL of the T7 clone into a 1.5 mL tubes, and add 5 units of DNase I
(2 µL) to each sample.
NOTE: The volume of phage sample selected at this step depends on the collected volume from biopanning. While the focus of the protocol is on the qPCR of biopanned phage, the steps for biopanning are detailed in the user protocol of the T7 phage kit (see Table of Materials)13.
- Option 2: Pipet 200 µL of the T7 clone into a 1.5 mL tubes, and add 5 units of DNase I
- Cap the T7 phage sample tubes (step 3.1) and incubate them at 37 °C for 10 min in a heated dry bath (i.e., heat block).
- Incubate tubes (from step 3.2) at 100 °C for 15 min in a heated dry bath (Figure 3A).
CAUTION: Since evaporation and condensation occur during the heating step, make sure that the 1.5 mL centrifuge tubes are capped completely. - Spin the heat-treated phages from step 3.3 at 18,534 x g for 10 s. Mix the phages by placing the tube on a vortex mixer set at touch-activation mode for 10 s, and spin at 18,534 x g for 10 s again.
NOTE: The spin-mix-spin cycle is to make sure the heated phage samples are well mixed. - Cool down the sample tubes at room temperature (RT) by leaving them on the experiment bench or store at 4 °C in a refrigerator overnight.
NOTE: Experiment can be paused at this step.
4. Prepare qPCR Reactions
NOTE: One PCR reaction is for one sample. Each PCR reaction contains 5 µL of qPCR master mix (see materials), 1 µL of 5 µM primer pair mix, 2 µL of H2O and 2 µL of heat-treated T7 phage sample. For multiple PCR reactions, all the reagents except phage sample are premixed in one 1.5 mL tube. The volume of each reagent in the premix depends on the number of phage samples for qPCR.
- Prepare the qPCR premix for 10 biopanned phage samples of unknown concentration and 7 samples of standard concentrations in triplicate. As a result, there are 51 total samples and therefore, 51 qPCR reactions (i.e., one reaction per sample). For 51 reactions, prepare a premix of 255 µL of qPCR master mix (51x 5 µL), 51 µL of primers (51x 1 µL), and 102 µL of H2O (51x 2 µL).
NOTE: It is recommended to prepare a few extra PCR reactions to compensate for the volume loss during aliquoting the PCR mix into each well of the 96-well qPCR plate. - Aliquot 8 µL of PCR premix into each well of 96-well qPCR plate for a total of 51 wells (i.e., 51 qPCR reactions). There is no need to change tips during aliquoting.
- Add 2 µL of heat-treated T7 phage samples (step 3.4) or T7 phage reference DNA (Step 2.1) to each well (Figure 3B); change tips when adding different DNA samples to the well to prevent cross-contamination. Also, dispense the 2 µL DNA samples underneath the surface of the qPCR premix (i.e., into the 8 µL PCR premix).
- Seal the qPCR plate with adhesive film.
NOTE: This step is critical, regardless of the type of qPCR plate. Use the recommended kit to seal the plate completely; otherwise, any non-sealed region in the plate will cause volume loss during PCR cycling. - Wrap the qPCR plate with aluminum foil to minimize fluorescence photobleaching and proceed to run it on the qPCR equipment.
5. qPCR Cycling Conditions
NOTE: qPCR cycling conditions were set up on the specific qPCR equipment (see Table of Materials).
- Run the 96-well qPCR plate on the qPCR equipment. On the equipment, select settings from the menu to set up the following experimental conditions (Figure 3C).
- Set up cycling conditions as follows for a sample volume of 10 µL: one cycle at 50 °C for 2 min, one cycle at 95 °C for 2 min, followed by 40 cycles of (95 °C for 15 s, 60 °C for 1 min). After, run the melt curve settings: one cycle of 95 °C for 15 s, 60 °C for 1 min, and 95 °C 15 s.
- Proceed to analyze the data as described in step 6.
6. Analyze the qPCR Raw Data and Convert from qPCR Ct Value to gc/µL for T7 Bacteriophages
- Analyze the raw qPCR data and evaluate data quality by analyzing the characteristic plots of the raw data (Figure 4).
NOTE: Amplification plot, multicomponent plot and melt curve plot are the key characteristic plots to validate the data quality of qPCR. - Calculate the mean threshold cycle (Ct) values from the triplicate samples at each concentration of the standard curve (step 2.1).
- Plot mean Ct (Y) against log (gc/µL) (X) to get the linear curve Y = kX + b (k is the slope of the linear curve, b is the intercept).
- Calculate the linear coefficient14 R2 (R2 must be >0.99).
- Use the standard curve Y = kX + b to calculate the number of T7 phages in the biopanning samples (i.e., phages selected against CF-like mucus) in gc/µL (Figure 5).
NOTE: k is the slope of the standard curve. - Calculate the amplification efficiency by the following equation
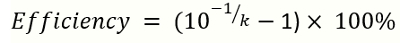 (2)
(2)
NOTE: k is the slope of the standard curve generated from reference DNA qPCR. The ideal range of amplification efficiency is 90–110% .
Different primer design tools can be used to design qPCR primers. Typically, primer design programs have their own built-in algorithms to calculate and validate the key parameters of the primers, e.g., GC%, Tm, primer dimer or hairpin formation, etc. Generally, the key criteria are similar in different primer design tools, and primers can be designed following their instructions. Primer BLAST can be used to confirm the specificity of the primers. One primer pair that targets upstream of the variable region of the T7 genomic DNA is shown in Figure 1. To determine the unknown concentrations of phage samples from biopanning, an absolute quantification approach – standard curve quantification – was chosen to enumerate the phage genomic DNA copies. Reference T7 phage DNA (see materials) at a known concentration 0.1 µg/µL was serially diluted from 1.0 x 100 to 1.0 x 106 fg/µL. Equation 1 was used to convert the T7 phage DNA concentrations from fg/µL to genome copies (gc)/µL; reference T7 phage DNA concentrations were 2.66 x 101 to 2.66 x 107 gc/µL (Figure 2).
After preparing dilutions of reference DNA for the standard curve, one selected CX7C T7 phage was prepared for qPCR (Figure 3A). Briefly, to describe the biopanning of the selected phage: an initial concentration of 4.2 x 109 pfu T7 CX7C phage was added on top of the apical compartment (donor) of a Transwell plate (see Table of Materials) containing 100 µL of CF-like mucus and 600 µL of phosphate buffered saline (PBS, see Table of Materials) in the basolateral compartment (receiver) and was incubated for 15 min at room temperature (25 °C). After 15 min, the eluate in the receiver was collected; from the eluate, one CX7C T7 clone was selected to be quantified by qPCR. qPCR reaction preparation was performed on ice with necessary reagents (e.g., qPCR master mix, primers, H2O), as shown in Figure 3B. The qPCR cycling conditions were set up on a qPCR thermocycler to run the samples (see Table of Materials, Figure 3C).
After the qPCR run, the amplification plot, melt curve plot and multicomponent plot (Figure 4) were analyzed from the raw data using the software (see Table of Materials). The amplification plot indicates the success or failure of PCR amplification of each sample. The multicomponent plot presents amplification and decay of fluorescence signal (reporter dye SYBR and background ROX dye) throughout the amplification cycles. The melt curve plot is used to validate the specificity of the primers since each PCR product has one unique melting temperature (Tm).
After evaluation of the qPCR raw data quality, the standard curve was generated from reference DNA: Ct values (Y) over log gc/µL (concentration conversion shown in Figure 5); here, it was Y = -3.5188 X + 35.09 (R2=0.999) (Figure 5A and 5B). The amplification efficiency was calculated to be 92.4%. The standard curve was used to interpolate the unknown concentrations of diluted T7 phage CX7C clone based on their Ct values. All reference samples and phage samples were run in triplicate, and their mean Ct values were used to calculate DNA concentration in gc/µL (Figure 5). The initial stock solution of the T7 clone was serially diluted to validate the stability and repeatability of qPCR for the enumeration of T7 phages5. At each dilution, samples were prepared in triplicate. The double-layer plaque assay was used as a reference method to determine the concentrations of the T7 dilutions; methods were described previously5. In Figure 6, the representative results from qPCR are shown compared to plaque assay at the tested range of 101 to 107 gc/µL. Titers or number of phages from qPCR quantitation were higher than from double-layer plaque assay. The repeatability of the qPCR method is represented by the coefficient of variance (CV) for concentrations from 101 to 107 gc/µL. The results were shown in Table 1; the CV% ranged from 2.85–27.2%.
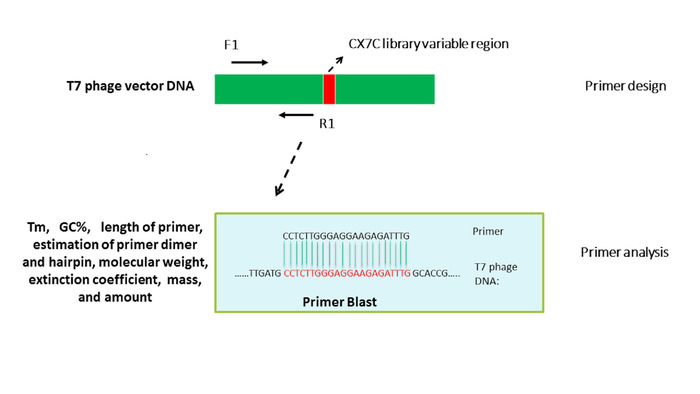
Figure 1: Primer design for T7-415 phage DNA vector and key parameters analysis of the designed primers. Oligo design tools were used to design multiple pairs of primers for T7-415 phage DNA. One set was selected after validation of the key parameters of the primers, e.g., Tm, GC%, primer-dimer and hairpin formation. Please click here to view a larger version of this figure.
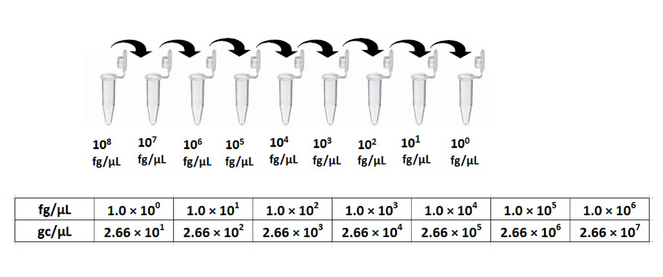
Figure 2: Standard curve preparation and unit conversion of T7 phage reference DNA. Serial 10-fold dilutions were made from 0.1 µg/µL T7 phage reference DNA for a standard curve. Equation 1 was used to convert DNA concentrations from fg/µL to gc/µL. Please click here to view a larger version of this figure.

Figure 3: Phage sample treatment, qPCR reaction preparation and qPCR run. DNase I pretreated T7 phage samples were heated at 100° C for 15 min to release the T7 DNA from intact phage particles (A); the qPCR mixture was prepared as described in the protocol. All the preparations were done on ice (B); qPCR equipment and compatible software (C) were used to set up the cycling conditions, run the PCR cycles, acquire, and analyze the raw data. Please click here to view a larger version of this figure.
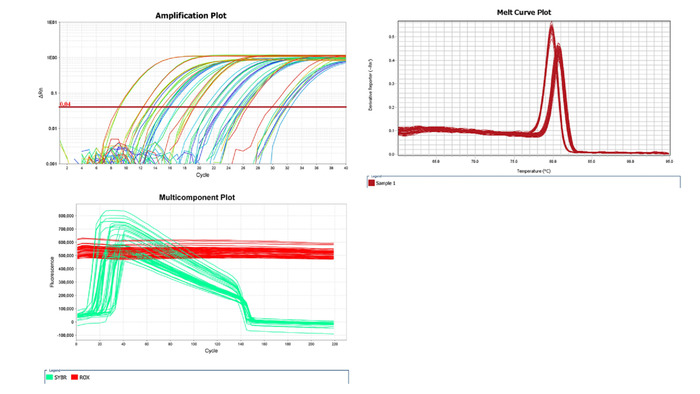
Figure 4: Characteristic plots of qPCR raw data. Amplification plot, multicomponent plot and melt curve plot were acquired from the qPCR raw data of one T7 phage CX7C clone from biopanning against in vitro CF-like mucus barrier. In the multicomponent plot, the passive reference dye (ROX) is a dye molecule included in the qPCR master mix that does not participate in the PCR amplification and will show no amplification signal in the plot. SYBR, which is SYBR green dye molecule in the qPCR master mix, will provide a fluorescence signal that should amplify and decay during qPCR cycling. Please click here to view a larger version of this figure.
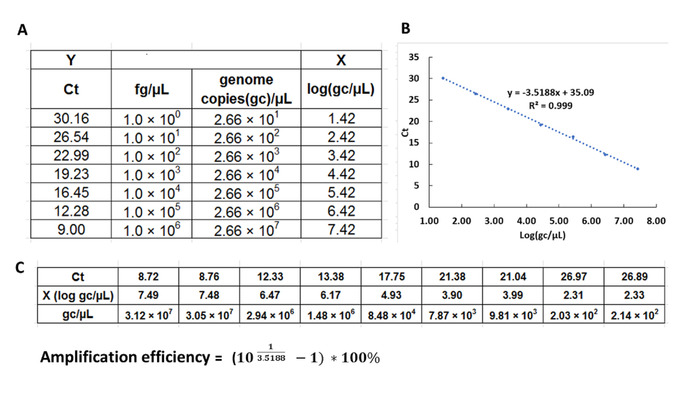
Figure 5: Analysis of raw data of qPCR to calculate gc/µL of the T7 phage particles. Threshold cycle (Ct) values of the standard curve of T7 phage reference DNA (Y) were plotted against logarithm transformation of the known concentrations of reference DNA in gc/µL (X) to get the standard curve (A and B). The standard curve was used to calculate the X (log gc/µL) values of the unknown concentrations of phage samples based on their Ct values. (C) T7 phage concentrations in gc/µL can be calculated. Also, calculation of qPCR amplification efficiency is provided. Please click here to view a larger version of this figure.
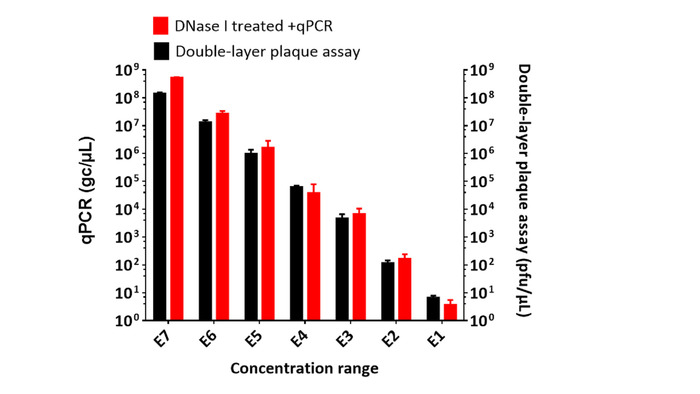
Figure 6: Sensitivity and repeatability results of qPCR to enumerate one T7 clone from biopanning against in vitro CF-like mucus barrier. One T7 CX7C phage clone selected from biopanning experiment was serially diluted into a concentration range (denoted as E1-E7 to represent 101 to 107) and in triplicate for qPCR and double-layer plaque assay. Both qPCR and double-layer plaque assay were used to quantify the concentration of the T7 phage clone at each dilution. Data were shown in mean value with the standard deviation (SD) at each scale for each treatment group. Please click here to view a larger version of this figure.
| Repeatability | |
| Concentration (gc/µL) | CV data in % |
| 10 1 | 27.2 |
| 10 2 | 7.65 |
| 10 3 | 2.85 |
| 10 4 | 15.7 |
| 10 5 | 15.6 |
| 10 6 | 5.03 |
| 10 7 | 4.71 |
Table 1: Repeatability of the qPCR method for one T7 phage CX7C clone. Intra-variability was tested at quantification scale of 101 to 107 gc/µL for the T7 clone DNA, the coefficient of variation (CV) was calculated as ratio of the standard deviation to the mean to express the repeatability of the qPCR method. CVs were computed in the range of 2.85–27.2% at for tested concentrations.
