Antigenic Liposomes for Generation of Disease-specific Antibodies
Summary
Described is the preparation of antigenic liposomal nanoparticles and their use in stimulating B-cell activation in vitro and in vivo. Consistent and robust antibody responses led to the development of a new peanut allergy model. The protocol for generating antigenic liposomes can be extended to different antigens and immunization models.
Abstract
Antibody responses provide critical protective immunity to a wide array of pathogens. There remains a high interest in generating robust antibodies for vaccination as well as understand how pathogenic antibody responses develop in allergies and autoimmune disease. Generating robust antigen-specific antibody responses is not always trivial. In mouse models, it often requires multiple rounds of immunizations with adjuvant that leads to a great deal of variability in the levels of induced antibodies. One example is in mouse models of peanut allergies where more robust and reproducible models that minimize mouse numbers and the use of adjuvant would be beneficial. Presented here is a highly reproducible mouse model of peanut allergy anaphylaxis. This new model relies on two key factors: (1) antigen-specific splenocytes are adoptively transferred from a peanut-sensitized mouse into a naïve recipient mouse, normalizing the number of antigen-specific memory B- and T-cells across a large number of mice; and (2) recipient mice are subsequently boosted with a strong multivalent immunogen in the form of liposomal nanoparticles displaying the major peanut allergen (Ara h 2). The major advantage of this model is its reproducibility, which ultimately lowers the number of animals used in each study, while minimizing the number of animals receiving multiple injections of adjuvant. The modular assembly of these immunogenic liposomes provides relatively facile adaptability to other allergic or autoimmune models that involve pathogenic antibodies.
Introduction
Food allergy affects 8% of children in the United States, and has increased in prevalence over the past decade1. Allergy to peanut affects 1% of children and is not typically outgrown2. Although several promising clinical trials are underway for the treatment of food allergy, including oral immunotherapy (OIT), sublingual immunotherapy (SLIT), and epicutaneous immunotherapy (EPIT), there are currently no FDA-approved treatment strategies for desensitizing peanut-allergic individuals3,4,5,6,7,8. Therefore, allergic individuals must strictly avoid allergens to avoid anaphylaxis. Many questions remain regarding routes of sensitization and underlying mechanisms of food allergy development.
Mouse models are a valuable tool for studying the mechanisms of allergy as well as developing new tolerogenic and desensitization therapies9,10,11,12. This is particularly true because the major peanut allergen (Ara h 2; Ah2) in humans is also the dominant allergen in several described mouse models13,14. While mouse models of peanut allergy are invaluable in studying mechanisms of sensitization and tolerance, a drawback is that they can be variable and require the use of adjuvants. More potent immunogens would be one way to minimize the intrinsic variability of such models. Since B-cells are strongly activated by multivalent antigens, antigenic liposomes displaying the allergen are a good option because of their ability to potentially activate B-cells through the B-cell receptor (BCR) while also having the property of efficiently priming the T-cell compartment through being taken up non-specifically by antigen-presenting cells.
Here, we describe a detailed protocol for conjugating protein antigens to liposomal nanoparticles using a facile and modular strategy. Using a surrogate antigen, anti-IgM Fab fragment, we demonstrate how potent such antigenic liposomes can be in stimulating B-cell activation. Antigenic liposomes displaying Ah2 antigen were used to develop a new mouse model of conferred sensitivity. In this model, splenocytes from verified peanut allergic mice, containing peanut-specific memory B- and T-cells, are transferred into naïve congenic mice. Memory antibody responses are induced by injection of liposomes conjugated with Ah2 into the recipient mice, in order to induce antibodies against Ah2. Followed by only one boost with soluble Ah2, Ah2-specific antibodies give rise to a strong anaphylactic response when these mice are subsequently challenged with Ah2. As mice undergoing the allergic reaction respond in a highly uniform manner and have not received an adjuvant, this approach is a desirable peanut allergy model and the outcomes suggest that it may have utility in other mouse models driven by antigens directed at allergens and possibly autoantigens.
Protocol
The general method of coupling protein to lipid and incorporating into liposomes is based largely on earlier work15. All animal procedures described below have been approved by the University of North Carolina at Chapel Hill Institutional Animal Care and Use Committee (IACUC). All mice used in the peanut allergy model are BALB/cJ females purchased from at 3 weeks of age. The University of Alberta Animal Care and Use Committee (ACUC) has approved experiments involving use of mouse spleens for ex vivo analysis from C57Bl/6 mice of at least 6 weeks of age.
1. Conjugation of Protein Antigen to PEGylated Lipid
- Add 3 g of cross-linked dextran gel beads with a fractionation range of 1,500–30,000 Daltons (Da) to a 250 mL vacuum flask. Add 60 mL of phosphate buffered saline (PBS) and continually stir with a magnetic stir bar. Seal the flask and attach to a vacuum to initiate de-gassing on a stir plate for a minimum of 1 h.
- In a 1.0 cm x 30 cm glass chromatography column with the turnover stopper closed, add PBS to the column to wash. Open the turnover stopper and drain the column. Following washing, add the slurry of de-gassed beads to the column. Continually add the slurry until the column is packed to a bead height of approximately 24 cm.
- Once the column has a ‘head’ of approximately 4–5 cm of PBS above the packed beads, have the following steps ready.
- Create a gravity flow siphon by measuring a piece of tubing long enough to reach a shelf 1–2 feet above the top of the column and create a loop that drops near the bottom of the column and returns to the top of the column. Add a turnover stopper to one end of the tubing, leaving one side of the tubing open. Place the open tube end into a 500 mL bottle of PBS and place on the shelf above the column.
- Take the end with the turnover stopper and attach a 10 mL syringe. Open the turnover stopper valve on the tubing line and draw PBS through the tube. Once the PBS has reached three quarters of the way through the tubing, quickly remove the syringe and add the turnover stopper to the top of the column — liquid should be running onto the column.
NOTE: The amount of ‘head’ of PBS left on the top of the column should be between 1 and 3 cm. The minimal amount to wash the column is 3 column volumes.
- Determine the amount of protein (moles) to be linked to lipid based on its molecular weight (MW).
NOTE: The Fab fragment of goat anti-mouse IgM (itself an IgG) is used here as a universal surrogate antigen for polyclonal stimulation of B-cells. Accordingly, the Fab fragment of goat IgG has a MW of approximately 48 kiloDalton (kDa) and is commercially available in a total quantity of 1.3 mg. Thus, the amount of protein to be linked is 27.1 µmol. - Determine the extinction coefficient of the protein of interest. If unknown, measure the protein absorbance at 280 nm using a spectrophotometer and divide the A280 value by the number of moles calculated in step 1.4.
NOTE: The extinction co-efficient of Fab of goat IgG at 280 nm is 64,600 M-1. - To ensure that trace amounts of amine-containing buffer are removed, desalt the protein on the column created in steps 1.1–1.3 and collect protein fractions into 1.5 mL microcentrifuge tubes (~500 µL per fraction).
- Close the turnover stopper valve connected to the top of the column and remove the top of the column. If not already open, open the turnover stopper valve at the bottom of the column and wait until the ‘head’ of PBS hits the top of the beads.
- Slowly add in the protein of interest to the top of the beads using a glass Pasteur pipette, being careful to minimize disturbance to the top of the beads.
- Once the ‘head’ of the protein solution has reached the top of the beads, add 1 mL of PBS gently via a Pasteur pipette. Repeat three times. Add PBS to create a head of 4–5 cm and repeat step 1.3 to wash the column.
NOTE: This step can be skipped if it is certain that no amine-containing buffer is present.
- Determine the fractions that contain protein by measuring A280 values and pool the fractions that give approximately 90% recovery of the protein.
NOTE: This typically dilutes the protein 1.5-2-fold. Determine the protein concentration after pooling. Concentrate the protein at this step, if necessary, using a centrifugation concentrator. - Add 2.5 molar equivalent of the heterobifunctional crosslinker to the protein.
- First, weigh approximately 5 mg of succinimidyl 3-(2-pyridyldithio)propionate (SPDP, MW = 314 g/mol) into a microcentrifuge tube. To determine the volume to dissolve the SPDP, take the molar protein concentration and multiply it by 250x to generate a 100x solution that will give a 2.5 molar excess in the reaction.
NOTE: For example, if the protein concentration is 50 µM, a solution of SPDP at 12.5 mM is needed. For 5 mg of SPDP, this corresponds to 1.27 mL of dimethyl sulfoxide (DMSO).
- First, weigh approximately 5 mg of succinimidyl 3-(2-pyridyldithio)propionate (SPDP, MW = 314 g/mol) into a microcentrifuge tube. To determine the volume to dissolve the SPDP, take the molar protein concentration and multiply it by 250x to generate a 100x solution that will give a 2.5 molar excess in the reaction.
- Add SPDP to the protein of interest at a 1:100 dilution. Place the reaction on an oscillating shaker at room temperature (RT) for approximately 1 h.
- To remove excess SPDP and protect the endogenous disulfide bond(s), in the protein in the subsequent reduction step, equilibrate, and wash the column created in steps 1.1–1.3 in 100 mM sodium acetate (NaOAc) at pH 5.5 or create an identical column solely for use in this buffer.
- After 1 h incubation with SPDP, desalt the protein on the column equilibrated in 100 mM NaOAc. Carry out this step in an identical manner as step 1.6 except using 100 mM NaOAc (pH 5.5) as the eluent. Collect the fractions, determine the A280, and pool the top fractions. Wash the column in PBS.
- Prepare a solution of 2.5 M of DTT (MW = 154 g/mol) in double distilled water (ddH2O). Add 25 mM DTT to the pooled fractions of protein for 5-10 min.
- Measure the A280 of the protein and divide this by the extinction coefficient. Also, determine the A343. Calculate the linking ratio based on the molarity of protein (A280) and linker (A343).
NOTE: Turn off the A340 auto-calibration if using a nanodrop spectrophotometer. The A343 measurement represents the absorbance of the pyridine 2-thione group (extinction coefficient = 7550 M-1) that is removed from the SPDP cross-linker by the DTT. A molar ratio of linker : protein ratio of 1.2–1.5 is generally achieved when performing the reaction with a 2.5 molar excess of SDPD on a wide range of proteins. - Run the pooled fractions over the column washed in PBS as described above. Collect the fractions, determine the A280, and pool top fractions. Determine the concentration of protein by A280 after pooling the fractions.
- Prepare DSPE-PEG(2000)-Maleimide (1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000)-Maleimide. This will be added to the protein at a 10:1 molar ratio. Prepare a 100x stock of DSPE-PEG(2000)-Maleimide to achieve the desired concentration.
- For example, if the protein concentration is 10 µM after step 1.14, prepare a stock of DSPE-PEG(2000)-Maleimide at 10 mM. Carefully weigh out DSPE-PEG(2000)-Maleimide (MW = 3000 g/mol) and add the appropriate amount of DMSO. Multiple rounds of gentle vortexing and sonicating may get it fully dissolved.
- Determine the total volume of the fractions from step 1.14 and transfer the solution carefully into a small (10-25 mL) round bottom flask (RBF). Add the appropriate amount of 100x DSPE-PEG(2000)-Maleimide to the protein to achieve a 1x, 10-fold molar excess, final concentration. Gently swirl the solution to ensure that the DMSO is fully dispersed.
- Run reaction overnight under nitrogen in a sealed RBF. Use a rubber septum and place in a fume hood.
- Remove the atmosphere by puncturing the septum with a 20 G needle attached to a 3 mL syringe at the end of tubing attached to a vacuum, and turn on vacuum for 3–5 s.
- Replace the atmosphere with nitrogen. Fill a balloon attached to a 3 mL syringe cut in half with nitrogen and attach a 20 G needle. Puncture the septum to fill the atmosphere inside the round bottom flask with nitrogen. Repeat the removal of the atmosphere and replacement with nitrogen once more and leave the nitrogen balloon attached overnight.
- Prepare a separate cross-linked dextran gel bead column with a fractionation range of 4,000–150,000 Da in a 1.0 cm x 50 cm glass chromatography column (as per steps 1.1-1.3).
- The following day, remove the protein from the round bottom flask prepared in step 1.17 and run (follow the method used in step 1.6) over the cross-linked dextran gel bead column (from step 1.18).
NOTE: The total amount to load onto the column should be no more than 2 mL. If a larger reaction was used, split into two and run on separate columns or run on a larger diameter column.- Collect the fractions, determine the A280, pool top fractions, and determine the concentration of protein. The presence of the lipid will not affect the measurements.
- Store the final pooled fractions of lipid-linked protein at 4 °C until ready for use.
2. Liposome Preparation
- Weigh out the lipids: DSPE-PEG(2000) (1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000, MW = 2805.5 g/mol; Cholesterol (3b-Hydroxy-5-cholestene, 5-Cholesten-3b-ol), MW = 386.7 g/mol; and DSPC (1,2-distearoyl-sn-glycero-3-phosphocholine), MW = 790.1 g/mol. Create a 5 mg/mL solution of DSPC, 4 mg/mL solution of Cholesterol, and 2 mg/mL solution of DSPE-PEG(2000) in chloroform.
- Ensure that the molar ratio of lipids in the liposomes is: 57% DSPC, 38% Cholesterol, and 5% DSPE-PEG(2000), as represented in Table 1.
NOTE: It is important to control the amount of antigen on the liposome, which generally ranges from 0.01–0.1%. In Table 1, 0.1% of goat anti-mouse IgM Fab fragment linked to PEGylated lipid is used. It is critical to take into account of the 10-fold molar excess of DSPE-PEG(2000)-Maleimide that was added to the protein in step 1.15, to ensure that the total amount of PEGylated lipid does not exceed 5%. - Consider the final lipid amounts (µmol) when creating lipids for extrusion. See Table 1 for an example of calculating molar lipid concentrations for the creation of liposomes at a 1.25 µM total lipid concentration.
- Once the correct amount of each lipid to be added together has been calculated, combine all into a 12 mL borosilicate glass test tube.
- Carefully blow off the chloroform.
- Use the tubing and a 3 mL syringe with needle, prepared in step 1.17.1, to carefully blow off chloroform in the tube with nitrogen. Gently blow a stream of nitrogen on the solution while rotating the tube with the other hand. Be careful to minimize splashing or causing the liquid to run way up the side of tube as it may be more difficult to get this into solution in the next step.
- Add 100 µL of DMSO to each tube and lyophilize this solution overnight. Cover the top of the tube with a non-static laboratory wipe, secure with a rubber band, and freeze -80 °C.
3. Liposome Extrusion
- Add 1 mL of PBS containing the lipid-linked protein to the 12 mL tube containing the lipids. Sonicate the solution for approximately 30 s to 1 min in a sonication water bath. Rest for 5 min or longer between each round and repeat 3–4 times.
- Prepare an extruder as directed by the manufacturer.
- Add the filter supports to the internal membrane support, place a few drops (10 µL each) of PBS on paraffin film. Using tweezers, grab an edge of the filter support and dip it in the 10 µL drop.
- Place the filter inside the O-ring. Do this for both sides. Using tweezers, grab a 0.8 µm filter on the edge and dip in the 10 µL drop and coat the outside of the O-ring with PBS. Place the 0.8 µm filter gently on the O-ring so the filter does not over hang the internal membrane support.
- Place the extruder heating block onto a hot plate to pre-heat the extruder. Switch the hot plate on low and allow the extruder heating block to reach the desired temperature. To avoid potential issues with protein aggregation, do not heat the block past 37 °C.
- Load the sonicated sample into one of the extruding syringes placed into the extruder. Place the extruder syringe into the other end of the extruder. Make sure the empty syringe plunger is set to zero. The empty syringe will fill as the lipid is extruded through the polycarbonate 0.8 µm membrane. Try to avoid passing bubbles back and forth through the membrane.
- Place the fully assembled extruder into the heating block. Gently push the plunger of the filled syringe with lipids to the empty syringe. Depending on the lipid concentration, this will be difficult to push through. Repeat 20x.
- Remove the fully assembled extruder from the heating block. Slowly remove the filled syringe from the extruder, be sure to collect any lipids that may leak out when removing. Place liposomes into a clean vial. Repeat steps 3.4–3.6 using polycarbonate 0.2 µm and 0.1 µm membranes.
- Create a 0.7 x 50 cm2 cross-linked agarose bead column with separation range of 0.7-10 Mega Daltons (MDa) equilibrated in PBS. Add the liposomes and run as in step 1.6. Fractions containing liposomes will have decreased transmission of light in the 250–400 nm range.
- Store the liposomes at 4 °C and do not freeze.
4. Calcium Flux to Monitor B-cell Activation by Antigenic Liposomes
- Euthanize mice by carbon dioxide (CO2) or isoflurane overdose, according to institutional standard operating procedures. Confirm euthanasia of mice by cervical dislocation.
- Sterilize surgical instruments and mice carcass using 70% ethanol. Use 10 cm long, 0.8 mm tip, curved iris forceps to separate the skin covering the chest cavity from the rib cage. Use 10 cm long, straight dissecting scissors to make a distal incision with a bilateral incision.
- Use curved iris forceps and dissecting scissors to extract the spleen from the abdominal cavity. Remove fatty tissue surrounding spleen and then place it in a 15 mL polystyrene conical tube filled with RPMI1640 medium containing 1% fetal calf serum (FCS), and penicillin-streptomycin.
- Transfer spleen and media over a 40 µm cell strainer fitted on a 50 mL conical tube. Using the rubber end of a 3 mL syringe plunger, gently crush the spleen. Wash the 40 µm cell strainer with the media. Repeat this process until only fat tissue is left in the 40 µm cell strainer. Rinse the sieve with 3-5 mL of media to increase cell recovery.
- Centrifuge cell homogenate at 300 x g for 5 min at RT. After centrifugation, discard the supernatant and add 10 mL of 1x RBC lysis buffer to the pellet. Pipette up and down, leave for 2–3 min at RT, and then centrifuge at 300 x g for 5 min at RT.
- Discard the supernatant and resuspend red blood cell depleted splenocytes in 10 mL of media. Determine total cell numbers using a hemocytometer or other cell counting device. Pellet the cells by centrifugation as described above.
NOTE: The ideal final concentration necessary for Indo-1 loading of splenocytes is 10–20 x 106 cells/mL, but a lower concentration of cells can be used. - Resuspend splenocytes at 15 x 106 cells/mL in calcium flux loading buffer (RPMI1640 medium containing 1% FCS, 10mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 1mM magnesium chloride (MgCl2), 1mM ethylene glycol-bis(β-aminoethyl ether)-N,N,N',N'-tetraacetic acid (EGTA), and 5% penicillin-streptomycin). Add in 1.5 µM Indo-1 from a 1 mM DMSO stock solution, invert the tube several times to mix. Protect from light, and incubate cells at 37 °C water bath for 30 min.
- Following the 30 min incubation, add in 5x the amount of calcium flux loading buffer. Centrifuge at 300 x g for 7 min at RT.
- For gating on B cells, stain cells with 1:200 anti-mouse CD5-PE and anti-mouse 1:200 B220-PE/Cy7 in 0.5 mL of loading buffer at 4 °C for 20 min, protected from light.
- Wash splenocytes in calcium flux loading buffer and centrifuge at 300 x g for 7 min at RT. Resuspend cells at 10–20 x 106 cells/mL in calcium flux running buffer (Hank’s balanced salt solution (HBSS) containing 1% FCS, 1 mM MgCl2 and 1 mM calcium chloride (CaCl2)). Store on ice, protected from light until ready to run on flow cytometer.
- Setup flow cytometry with unstained and single stain controls for compensation.
- Run the stained cells to setup the gates appropriately (see Figure 2A). Setup Indo-1 (violet) versus Indo-1 (blue) plot, adjust the voltages in the Indo-1 channels to place the cells staining at a slope of 45–60° to maximize the signal : noise ratio of the Indo-1 violet : blue change in ratio following B-cell stimulation.
NOTE: Make sure that the voltages are not too high, such that a significant percentage of the cells (>5%) are up against the top of the plot. - Create a diagonal gate that encapsulates the top left portion (Violet+Blue–) of the plot, such that without stimulation <10% of the cells are in the gate, which should ideally increase to >75% following stimulation.
- Run the stained cells to setup the gates appropriately (see Figure 2A). Setup Indo-1 (violet) versus Indo-1 (blue) plot, adjust the voltages in the Indo-1 channels to place the cells staining at a slope of 45–60° to maximize the signal : noise ratio of the Indo-1 violet : blue change in ratio following B-cell stimulation.
- Add 0.5 mL of cells to a capped 5 mL round bottom polystyrene tube (fluorescence-activated cell sorting (FACS) tube) and warm the cells to 37 °C for 3–5 min in a water bath. Place the FACS tube in the 37 °C water-jacketed chamber that is connected to a re-circulating water bath.
- Run the tube in the water-jacket on the flow cytometer and initiate acquisition, without storing the data, at approximately 5,000–10,000 events/s. Once the cells have stabilized (15–30 s), initiate data acquisition and collect data for at least 10 s to establish the background.
- At the 10 s mark, quickly remove the tube from the flow cytometer, add the stimulation (5-50 μM antigenic liposomes in 2–20 μL), pulse vortex, and return FACS tube onto the flow cytometer. Maintain data acquisition and storage through this time and collect data for 3–5 min.
- Analyze data in appropriate analysis software under the kinetics functions.
5. Preparation of Peanut Extract
- Extract peanut proteins by mixing peanut flour in a 1:5 (wt:vol) ratio of phosphate buffered saline (PBS) with 1 M sodium chloride (NaCl). Mix solution on magnetic stir plate for 2 h at RT while maintaining at pH 8.5.
- Centrifuge solution at 3,000 x g for 45 min at 4 °C. Decant and filter-sterilize supernatant sequentially through a 0.4 µm filter followed by a 0.2 µm filter. Determine protein concentration by bicinchoninic acid (BCA) assay using bovine serum albumin (BSA) as the standard.
- Reduce and denature a sample of peanut proteins with 50 mM dithiothreitol (DTT) in a buffer containing lithium dodecyl sulfate at a pH of 8.4, which allows for maximal activity of the reducing agent and heat at 70 °C for 10 min.
- Run 10 µg of denatured peanut protein on a Bis-Tris 4–12% precasted polyacrylamide gel designed to give optimal separation gradient gel with molecular weight pre-stained standard.
- Stain the gel with disulfonated triphenylmethane and destain with ddH2O. Identify known major peanut allergens Ara h 1, 2 and 3 in extract preparations. Ara h 1 appears at 63 kDa, Ara h 2 should appear as two isoforms at 17 and 19 kDa, and Ara h 3 appears at 37 kDa (acidic subunit).
6. Sensitization of Mice to Peanut
- Resuspend 1 mg Cholera toxin in 1 mL ultrapure water. Prepare 200 µL diluted peanut extract containing 2 mg peanut protein and 10 µg cholera toxin in PBS.
- Administer 200 µL of the peanut extract containing Cholera toxin (2 mg peanut protein and 10 µg Cholera toxin) via oral gavage to each 4–5 week old BalB/cJ female mouse. Repeat once a week for three weeks (i.e., Days 0, 7, and 14).
- Administer 300 µL diluted peanut extract (containing 5 mg peanut protein and 10 µg Cholera toxin) via oral gavage to each mouse in the fourth week (i.e., Day 21).
7. Challenge Mice with Peanut Extract
- On day 28, prepare peanut extract to a final concentration of 1 mg/mL in PBS. Measure baseline body temperatures using a rectal thermometer (approximately 37.5–38.5 °C). Administer 200 µL (200 µg) of peanut extract via intraperitoneal (i.p.) injection.
- Measure body temperature with rectal thermometer every 15 min for 1 h after injection. A dip in body temperature indicates allergy to peanut.
NOTE: Hypothermia is a hallmark of anaphylaxis in mice. This challenge will demonstrate that mice are allergic to peanut. Typically, 90% of Balb/cJ mice that undergo the sensitization protocol develop allergic reactions during challenge characterized by at least a 2–3 °C temperature decrease.
8. Isolation of Splenocytes from Allergic Mice and Adoptive Transfer
- Isolate the splenocytes from naïve and peanut allergic mice.
- On day 30, euthanize both naïve and confirmed peanut allergic mice by administering 3 L/min of CO2 in a gas chamber. Confirm euthanasia of mice by bilateral thoracotomy. Sterilize surgical instruments and mouse carcass using 70% ethanol. Use forceps to separate the skin covering the chest cavity from the rib cage. Use straight dissecting scissors to make a bilateral incision breaking the left and right ribs.
- Use forceps and dissecting scissors to extract the spleen from the abdominal cavity. Remove fatty tissue surrounding spleen before placing in polystyrene 15 mL conical tube, filled with 10 mL RPMI 1640 medium.
- In a sterile laminar flow hood, pour spleen and media into a 35 mm polystyrene Petri dish. Keep naïve and allergic splenocytes in separate Petri dishes and media during cell isolation. Use sterilized forceps and dissecting scissors to cut spleen into three pieces and return to Petri dish.
- Using the rough, frosted edge of two glass microscope slides, gently homogenize (in RPMI) the spleen pieces until white tissue remains between slides. Change slides between groups.
- Transfer cell homogenate from Petri dish through a 70 µm cell strainer fitted on a 50 mL conical tube. Pass the remaining spleen fragments through the strainer with a plunger from a 1 mL syringe. Rinse the sieve with 5 mL of RPMI to maximize cell recovery. Transfer filtrate to a polystyrene 15 mL conical tube, and centrifuge (450 x g, 10 min, RT).
- Aspirate supernatant and add resuspend cell pellet in 2 mL red blood cell lysis buffer (0.83% ammonium chloride in 10 mM Tris buffer, pH 7.2). Mix and incubate at RT for 2–3 min. Add 10 mL of PBS-2% FBS and centrifuge at 300 x g for 7 min at RT.
- Wash the cells again with 12 mL PBS-2% FBS to completely remove any residual ammonium chloride, and centrifuge at 300 x g for 7 min at RT. Re-suspend cell pellet in 2 mL of RPMI 1640 medium. Count cells using either a hemocytometer or automatic hematology analyzer.
- Using a 27 G, 5/8-inch needle on a 1 mL insulin syringe, intravenously inject 15 x 106 allergic or naïve splenocytes extracted as described above (in 200 µL) via the tail vein into naïve (unsensitized) mice.
9. Injection of Mice with Ara h 2 Antigenic Liposomes
- Prepare Ah2 liposomes containing 0.1% Ah2 at 2.5 mM lipid. For calculating protein concentration, the extinction coefficient of Ah2 is 15,000 M-1. Dilute liposomes to 300 µM in sterile PBS.
- One day after adoptive transfer (day 31) of allergic or naive splenocytes, intravenously inject 200 µL of PBS or the Ah2 antigenic liposomes (300 µM) containing a total of 1.38 µg of Ah2 via the tail vein of BALB/cJ mice that received the splenocytes in step 8.2.
10. Boost and Challenge of Mice with Ara h 2
- Two weeks after the injection with Ah2 liposomes, on day 45, use a 4 mm animal lancet to collect 50–200 µL of blood from the submandibular junction of the mouse. Collect blood in a serum separator tube without heparin; separate the serum by centrifuging at 6,000 x g for 15 min. Use the collected serum later on to measure antigen-specific antibodies.
- On day 46, two weeks after injecting mice with liposomes (step 9.2), boost mice by an i.p. injection of 200 µL of PBS or 200 µg of soluble Ah2 (purified as described previously16).
- On day 60, collect 50-200 µL of blood from the submandibular junction of the mouse, and process blood as described previously.
- On day 61, challenge each group of mice with an i.p. injection of 200 µL of 300 µg soluble Ah2. Measure body temperatures with rectal probe every 15 min as in Section 7.
11. Quantification of Ara h 2-specific IgE and IgG1 by ELISA
- Coat a 96-well enzyme-linked immunosorbent assay (ELISA)-compatible plate with 100 µL HSA-DNP (2,4-dinitrophenyl hapten conjugated to human serum albumin, 20 µg/mL) for standard curve or Ah2 (5 µg/mL) for serum samples collected in steps 10.1 and 10.3, diluted in coating buffer (50 mM carbonate-bicarbonate buffer, pH 9.6) at 4 °C overnight or 37 °C for >1 h.
- Wash three times with 200 µL PBS-T (PBS with 0.05% Tween-20) and block wells with 200 µL PBS-T containing 2% BSA at 37 °C for >2 h.
- For Ah2-specific IgE ELISA, add 50 µL IgE-anti-DNP standards (2–0.002 µg/mL, prepared from 1:2 serial dilutions) or mouse serum samples (1:20 dilution) and incubate overnight at 4 °C. For Ah2-specific IgG1 ELISA, add 100 µL purified IgG1-anti-DNP standards (2–0.002 µg/mL, prepared from 1:2 serial dilutions) or mouse serum samples (1:500 dilution) and incubate overnight at 4 °C or 1 h at 37 °C.
- Wash three times with 200 µL PBS-T. For Ah2-specific IgE ELISA, add 100 µL Sheep anti-Mouse IgE (0.5 µg/mL) and incubate at 37 °C for 1 h. For Ah2-specific IgG1 ELISA, add 100 µL HRP Goat anti-Mouse IgG1 (diluted 1:40,000 in PBS-T-2% BSA). Incubate at 37 °C for 1 h.
- Wash three times with 200 µL PBS-T. For Ah2-specific IgE ELISA, add 100 µL biotinylated-Donkey anti-Sheep IgG (0.5 µg/mL) and incubate at 37 °C for 45 min. For Ah2-specific IgG1 ELISA, skip to step 11.7.
- Wash three times with 200 µL PBS-T. For Ah2-specific IgE ELISA, add 100 µL neutrally charged avidin-horseradish peroxidase (e.g., NeutrAvidin-HRP, 0.3 µg/mL) and incubate at 37 °C for 30 min.
- Add 100 µL 3,3’,5,5’-Tetramethylbenzidine (TMB) substrate, incubate 10-15 min, or until samples turn dark blue, then add 100 µL TMB stop solution. Read the plate at 450 nm.
Representative Results
Conjugation of the protein of interest with DSPE-PEG(2000) can be demonstrated by running a reducing showing an increase in molecular weight compared to the unconjugated protein. Figure 1A shows a representative gel of anti-mouse IgM F(ab) fragment conjugation to PEG-DSPE, which shows a 2–3 kDa bandshift for the denatured protein. Note that approximately 50% of the protein appears to be modified, which is expected given that 1:1 stoichiometry was achieved on the Fab fragment that is a heterodimer of the heavy and light chain. Figure 1B shows a representative gel of Ah2 conjugation to PEG-DSPE. To assess calcium flux of B-cells stimulated by antigenic liposomes, two things are crucial: (1) the instrument settings are tuned to see a difference in the ratio of Indo-1 fluorescence in the Ca2+ bound (violet) and unbound (blue) forms and (2) the proper gating strategy is used to assess B-cell activation. Figure 2A shows the gating scheme for the flow cytometry-based calcium flux assay. Live lymphocytes are gated in an SSC-A versus FSC-A (left panel), doublets are gated out in a FSC-W versus SSC-W plot (middle panel), and B200+CD5– B-cells are selected from a CD5-PE vs B220-PE/Cy7 plot (right panel). Figure 2B demonstrates the ratio of Indo-1 (violet) vs Indo-1 (blue) fluorescence over time as analyzed. Note that the total protein concentration of F(ab) and F(ab')2 in these assays were the same, demonstrating the superior ability of antigenic liposomes in stimulating B-cell activation. After preparing the peanut extract, run an aliquot on an SDS-PAGE gel to determine the relative quantities of Ara h 1, 2 and 3 within the extract. Figure 3 shows a representative gel of a peanut extraction that was run alongside purified Ah2. Figure 4 shows a schematic of the overall adoptive peanut allergy mouse model, including initial sensitization to peanut, challenge to peanut, splenocyte isolation and adoptive transfer, liposome injections, blood draw followed by Ah2 boost, and challenge to Ah2. Ah2-specific IgE and IgG1 ELISAs were run to quantify immunoglobulins in serum, as shown in Figure 5A and B. Mice with conferred sensitivity that have been boosted with Ah2 will have Ah2-specific IgE and IgG1 in their serum. Body temperatures recorded during the Ah2 challenge are shown in Figure 5C; allergic mice had decreased body temperatures following the challenge, while body temperatures in naïve mice remained consistent. Photos showing naïve mice compared to peanut-allergic mice during the challenge are shown in Figure 6.
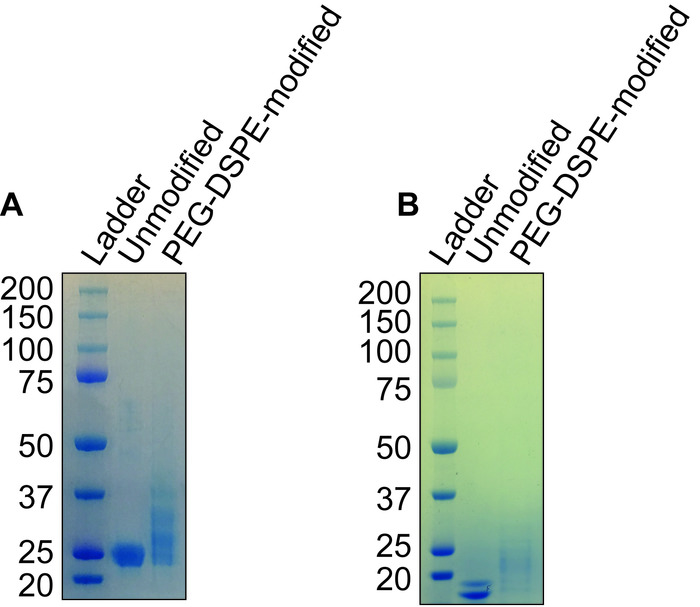
Figure 1: Representative gel of anti-mouse IgM F(ab) and Ah2 conjugation to PEG-DSPE. (A, B) SDS-page analysis of (A) goat anti-mouse IgM F(ab) fragment and (B) Ah2 before and after conjugation to PEG-DSPE. Goat anti-mouse IgM and Ah2 have molecular weights of approximately 48 and 18 kDa, respectively. Following modification, a slightly larger molecular weight (~2.5 kDa) is clearly observed for both proteins. Please click here to view a larger version of this figure.
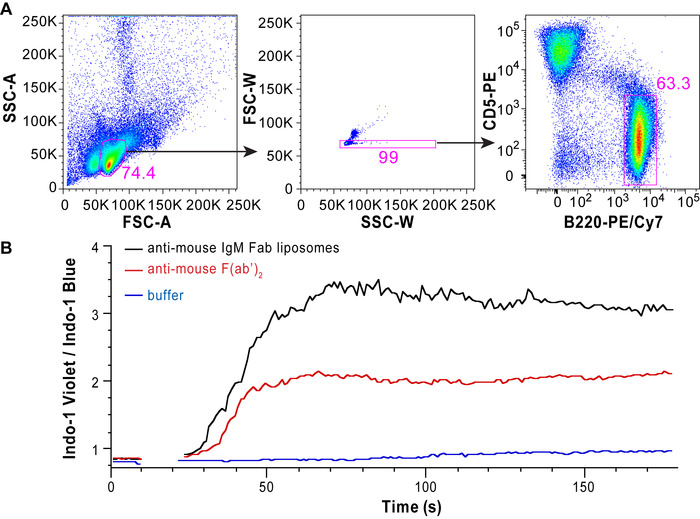
Figure 2: Representative gating strategy, calcium flux results, and analysis. (A) Cells were analyzed through the following gating strategy: live lymphocytes (FSC-A vs. SSC-A), single cells (FSC-W vs. SSC-W), and B-cells (B220+CD5–). (B) The Indo-1/Ca2+ flux response (violet vs. blue) of the B-cells. Shown is the calcium flux for anti-IgM Fab fragment liposomes and anti-IgM F(ab')2 at the same protein concentration (2.5 µg/mL) as well as buffer-stimulated cells as a control. Note that between 10-22 s is when the stimulation is added and, therefore, no data is acquired during this time. Please click here to view a larger version of this figure.
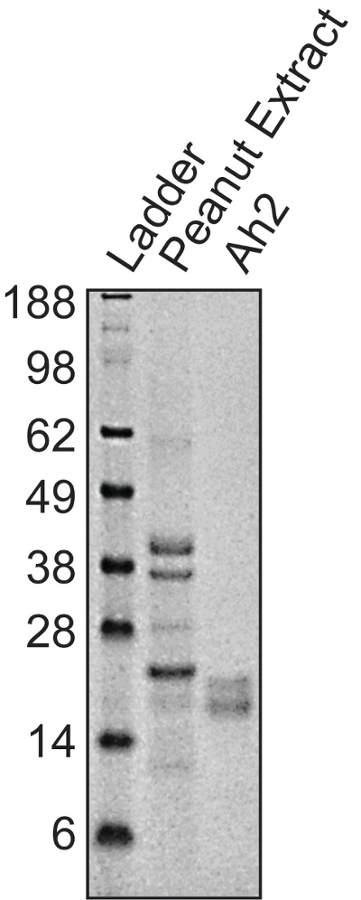
Figure 3: Representative gel showing peanut extract and Ah2. Peanut extract contains several proteins, including allergens Ara h 1, 2, 3 and 6, compared to the two isoforms that appear for Ah2. Please click here to view a larger version of this figure.
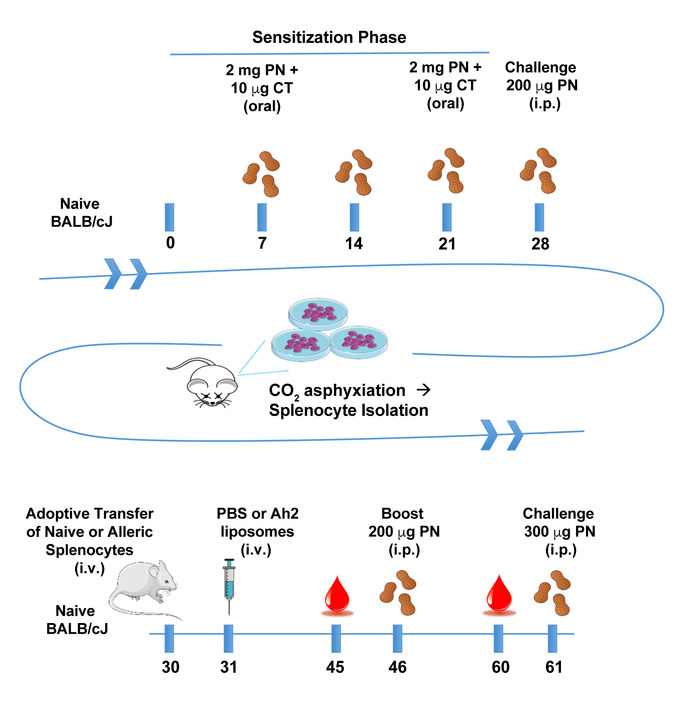
Figure 4: Schematic of adoptive transfer protocol. Naïve BALB/cJ mice were sensitized with peanut extract (PN) and cholera toxin (CT), and subsequently challenged to PN. Splenocytes from confirmed allergic mice were isolated and transferred into naïve mice. Mice were later primed with immunogenic Ara h 2 liposomes or PBS, then boosted with soluble Ah2. Finally, mice were challenged with Ah2 to monitor anaphylaxis. Blood collected on days 45 and 60 were used to quantify Ah2-specific immunoglobulins. Please click here to view a larger version of this figure.
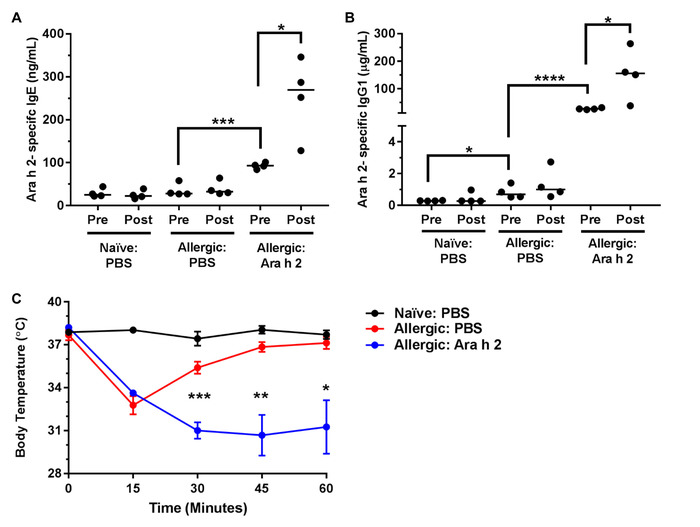
Figure 5: Immunogenic Ara h 2 liposome boosts conferred-memory allergic responses. Serum was isolated pre (day 45) and post (day 60) boost with PBS or 200 µL of 300 µM immunogenic Ah2 liposome to measure Ah2-sepcific IgE (A) and IgG1 (B). Individual mice are represented with lines indicating medians. Mice that received allergic splenocytes and were administered Ah2 liposomes on day 32 had significantly higher levels of Ah2-specific IgE and IgG1. Anaphylaxis was measured in the different treatment groups by recording body temperatures after Ah2challenge (C). Mean body temperatures depicted with SEM. Naïve: PBS (Naïve splenocytes and PBS prime), Allergic: PBS (confirmed allergic splenocytes and PBS prime), Allergic: Ah2 (confirmed allergic splenocytes and Ah2immunogenic liposome prime). * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001 determined by unpaired 2-tailed Student's t-test. Note that these results are representative of two independent experiments. Please click here to view a larger version of this figure.
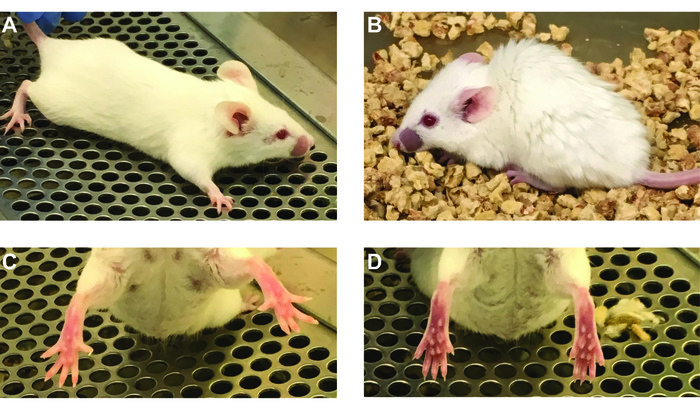
Figure 6: Anaphylactic symptoms observed during Ah2 challenge. Naïve mice remain active and have pink complexion on their feet (A, C). Allergic mice have decreased activity, are often hunched up, have labored breathing and experience cyanosis, indicated by darker purple complexion on their feet (B, D). Please click here to view a larger version of this figure.
| MW | 790 | 387 | 2900 | 3000 | 48000 | |
| DSPC | Cholesterol | PEG-DSPE | excess PEG-DSPE | aIgM-PEG-DSPE | TOTAL | |
| Molar ratio | 57 | 38 | 3.9 | 1 | 0.1 | 100.00 |
| mass (mg) | 0.56 | 0.18 | 0.14 | 0.04 | 0.06 | 0.98 |
| mmol | 0.71 | 0.47 | 0.05 | 0.01 | 0.00 | 1.25 |
| mL | 112.50 | 45.93 | 70.64 | 0.00 | 525.97 | 755.03 |
| Conc. (mg/mL) | ||||||
| DSPC | 5 | |||||
| Cholesterol | 4 | |||||
| PEG-DSPE | 2 | |||||
| aIgM-PEG-DSPE | 0.114 |
Table 1: Calculations for creating a 1.25 µM total lipid concentration liposome. Example of calculation table for goat anti-mouse IgM F(ab) fragment liposomes.
Discussion
The methods outlined here are a general protocol for the conjugation of a protein to a lipid which enables the display of the protein on liposomal nanoparticles. For very large multi-subunit proteins, this protocol may have limited utility. The ideal method would be the introduction of a site-specific tag that enables a biorthogonal chemical linking strategy to be used. If expressing the protein recombinantly, this can be possible using available site-specific strategies17, and a wide array of functional groups at the end of the PEGylated lipid that are commercially available. Accordingly, this protocol is geared towards the linking of protein isolated from natural sources and is accessible to a wide-range of scientists not necessarily familiar with chemical and biochemical transformations.
A key aspect of running a calcium flux experiment that should to be emphasized is that the cells should not be activated before running them on the flow cytometry. If sterile technique is not rigorously used, this will account for a high background signal from activated cells (Indo-1 fluorescence skewed towards its emission in the violet channel). Similarly, cells should be kept on ice prior to running the assay to avoid abnormal levels of background signal in the Indo-1 channels. It is also important to ensure that the cells are warmed for 2–3 min prior to acquiring by flow cytometry and throughout stimulation during data acquisition. If the cells are not maintained at 37 °C during data acquisition, it can be expected that cells will not respond maximally. Since antigen-specific B-cells are present in mice at very small numbers, using a surrogate antigen to stimulate all B-cells from mice is ideal. Although numerous transgenic mouse strains are available that express antigen-specific B-cells, the purpose of this technique is to enable the average user to be able to validate their ability to make antigenic liposomes using wild-type mice. To accomplish this objective, Fab fragments of anti-mouse IgM were linked to lipids and formulated into antigenic liposomes that cross-link the B-cell receptor (BCR) in a polyclonal fashion. It is noteworthy that it was previously demonstrated that this method can be used to stimulate human B-cells10 using the appropriate anti-human IgM Fab fragment, which is significant because it is not possible to access significant numbers of antigen-specific human B-cells. In these studies, the potency of antigenic liposomes to stimulate B-cell activation was illustrated by their enhanced ability to induce calcium flux compared to equal amounts of anti-mouse IgM F(ab')2 fragments.
The use of antigenic liposomes has enabled the development of a method for sensitizing BALB/cJ mice to peanut, transferring splenocytes from allergic mice into naïve mice, injecting the host mice with Ah2 liposomes, and challenging them with Ah2. Antibody levels and anaphylactic responses within groups of mice are reproducible in this model. It has been well-established that memory B- and T-cell responses are critical in the development of peanut-specific antibodies in mice. Moreover, a recent study has shown that memory B-cells repopulate plasma cells in mice, which maintain elevated antigen-specific IgE levels; therefore allergen-specific memory B-cells are an attractive target for therapeutic intervention18. The model described here now allows an opportunity to directly target Ah2-specific memory B cells, as opposed to an immunosuppressive approach using bortezomib to deplete the entire plasma cell repertoire19. Another approach to combat allergies is to induce tolerance in the T-cell compartment. One potential strategy is to encapsulate tolerance-inducing adjuvants within the liposome. Previously, nanoparticles encapsulating rapamycin were used to induce Tregs and diminish autoimmune disease20. This type of approach could be employed to develop tolerogenic therapies to treat peanut allergy. Overall, manipulation of the allergen-specific liposome by conjugating molecules to the surface allows for tailored immune responses, and encapsulating drug-like molecules enables delivery of drug to the allergen-specific cell.
General limitations of this mouse model of peanut allergy are that sensitization to peanut with Cholera toxin and subsequent peanut challenge via i.p. injection is not entirely representative of peanut allergy in humans (e.g., humans have reactions upon oral ingestion). However, this allergy model is the current standard in the field11,18,21, and allows us to investigate molecular mechanisms of allergic disease and develop novel therapeutic approaches. Compared to these conventional approaches, this adoptive transfer model offers three primary advantages. The first is that it enables memory B- and T-cells specific to the allergen to be studied more directly. Indeed, evidence now strongly implicates memory B-cells as the major source of long-term allergies18. Second, because the same number of memory B- and T-cells are implanted into each recipient mouse, the allergic responses are minimally variable. Third, this model provides the opportunity to study allergic responses to individual allergens, which is useful in the context of testing new immunotherapies. One intriguing application of this model is to use the general approach in the context of a humanized mouse model, where allergic responses have been studied in immunized models22. Applied in the context of an adoptive transfer model, a humanized mouse model could be a powerful approach to expanding and in vivo testing of B- and T-cells from allergic patients.
Antibody disease pathogenesis does not only occur in allergic models, but also in B-cell mediated autoimmune diseases23. Adoptively transferring sensitized splenocytes from mice given an autoantigen would ultimately be very useful in lowering the number of animals used, minimize the number of animals receiving multiple injections of adjuvant, and increase the reproducibility of responses between cohorts of mice in multiple disease indications. One example would be in an experimental autoimmune model of myasthenia gravis (EAMG) in which susceptible mouse strains (C57BL/6, SJL, and AKR) are immunized multiple times with acetylcholine receptor (AChR) in adjuvant to develop pathogenic autoantibodies. These autoantibodies ultimately lead to immunopathological features present in humans such as muscle fatigue/weakness, deposit of immunoglobulins, and complement components on neuromuscular junctions and in severe cases morbidity/death24. In EAMG, the disease incidence varies widely between 50–70%, so to account for this variability large animal cohorts are needed to reach statistical significance25,26. The method described here would ultimately reduce the number of animals used by decreasing the variability in disease incidence. As previously mentioned, EAMG is induced by multiple injections with AChR + adjuvants (Complete Freund's Adjuvant and Incomplete Freund's Adjuvant) to trigger auto-antibody responses. This method would reduce the number of animals that receive multiple injects with adjuvant. Finally, due to the nature of multiple immunizations and boosts, it is relatively difficult to define appropriate therapeutic windows for prophylactic and therapeutic treatments. This method would help synchronize the antibody response and disease pathogenesis window, making it a more predictable and reproducible. This same logic can be applied to many other mouse models of antibody mediated autoimmune diseases, such as Rheumatoid arthritis27.
Disclosures
The authors have nothing to disclose.
Acknowledgements
This research was supported by grants from the Department of Defense (W81XWH-16-1-0302 and W81XWH-16-1-0303).
Materials
| Model 2110 Fraction Collector | BioRad | 7318122 | |
| Cholestrol | Sigma | C8667 | Sigma grade 99% |
| SPDP | Thermo Fisher Scientific | 21857 | |
| DSPC | Avanti | 850365 | |
| DSPE-PEG 18:0 | Avanti | 880120 | |
| DSPE-PEG Maleimide | Avanti | 880126 | |
| Extruder | Avanti | 610000 | 1mL syringe with holder/heating block |
| Filters 0.1 µm | Avanti | 610005 | |
| Filters 0.8 µm | Avanti | 610009 | |
| 10mm Filter Supports | Avanti | 6100014 | |
| Glass Round Bottom Flask | Sigma | Z100633 | |
| Turnover stoppers | Thermo Fisher Scientific | P-301398 | |
| Tubing | Thermo Fisher Scientific | P-198194 | |
| Leur Lock | Thermo Fisher Scientific | k4201634503 | |
| Sephadex G50 Beads | GE Life Sciences | 17004201 | |
| Sephadex G100 Beads | GE Life Sciences | 17006001 | |
| Heat Inactivated Fetal Calf Serum | Thermo Fisher Scientific | 10082147 | |
| HEPES (1M) | Thermo Fisher Scientific | 15630080 | |
| EGTA | Sigma | E3889 | |
| Penicillin-Streptomycin (10,000 U/mL) | Thermo Fisher Scientific | 15140122 | |
| 1x RBC lysis Buffer | Thermo Fisher Scientific | 00-4333-57 | |
| Indo-1 | Invitrogen | I1203 | |
| CD5-PE | BioLegend | 100608 | |
| B220-PE-Cy7 | BioLegend | 103222 | |
| HBSS | Thermo Fisher Scientific | 14170112 | without calcium and magnesium |
| MgCl2 | Sigma | M8266 | |
| CaCl2 | Sigma | C4901 | |
| Fab anti-mouse IgM | Jackson ImmunoResearch | 115-007-020 | |
| F(ab')2 anti-mouse IgM | Jackson ImmunoResearch | 115-006-020 | |
| Peanut flour | Golden Peanut Co. | 521271 | 12% fat light roast, 50% protein |
| Animal feeding needles | Cadence Science | 7920 | 22g x 1.5", 1.25 mm – straight |
| Microprobe thermometer | Physitemp | BAT-12 | |
| Rectal probe for mice | Physitemp | Ret-3 | |
| Cholera toxin, from vibrio cholera | List Biological Laboratories, Inc. | 100B | Azide free |
| BCA Protein Assay Kit | Pierce | 23225 | |
| Carbonate-bicarbonate buffer | Sigma | C3041 | |
| TMB Stop Solution | KPL | 50-85-06 | |
| SureBlue TMB Microwell Peroxidase Substrate | KPL | 5120-0077 | |
| 96 well Immulon 4HBX plate | Thermo Scientific | 3855 | |
| Purified soluble Ara h 2 | N/A | N/A | purified as in: Sen, et al., 2002, Journal of Immunology |
| HSA-DNP | Sigma | A-6661 | |
| Mouse IgE anti-DNP | Accurate Chemical | BYA60251 | |
| Sheep anti-Mouse IgE | The Binding Site | PC284 | |
| Biotinylated Donkey anti-Sheep IgG | Accurate Chemical | JNS065003 | |
| NeutrAvidin Protein, HRP | ThermoFisher Scientific | 31001 | |
| Mouse IgG1 anti-DNP | Accurate Chemical | MADNP105 | |
| HRP Goat anti-mouse IgG1 | Southern Biotech | 1070-05 | |
| 1 mL Insulin Syringes | BD | 329412 | U-100 Insulin, 0.40 mm(27G) x 16.0 mm (5/8") |
| Superfrost Microscope Slides | Fisher Scientific | 12-550-14 | 25 x 75 x 1.0 mm |
| ACK Lysing Buffer | gibco by Life Technologies | A10492-01 | 100 mL |
| RPMI 1640 Medium | Thermo Fisher Scientific | 11875093 | 500 mL |
| Cell Strainer | Corning | 352350 | 70 μm Nylon, White, Sterile, Individually packaged |
| NuPAGE 4-12% Bis-Tris Protein Gels | Invitrogen | NP0322BOX | 10 gels |
| NuPAGE LDS buffer, 4X | Invitrogen | NP0008 | 250 mL |
| SeeBlue Plus2 Pre-stained standard | Invitrogen | LC5925 | 500 µL |
| NuPAGE MES/SDS running buffer, 20X | Invitrogen | NP0002 | 500 mL |
| GelCode Blue Stain | Thermo Scientific | 24590 | 500 mL |
References
- Gupta, R. S., et al. The prevalence, severity, and distribution of childhood food allergy in the United States. Pediatrics. 128 (1), e9-e17 (2011).
- Sicherer, S. H., Munoz-Furlong, A., Godbold, J. H., Sampson, H. A. US prevalence of self-reported peanut, tree nut, and sesame allergy: 11-year follow-up. Journal of Allergy and Clinical Immunology. 125 (6), 1322-1326 (2010).
- Kim, E. H., et al. Sublingual immunotherapy for peanut allergy: clinical and immunologic evidence of desensitization. Journal of Allergy Clinical Immunology. 127 (3), 640-646 (2011).
- Vickery, B. P., et al. Sustained unresponsiveness to peanut in subjects who have completed peanut oral immunotherapy. Journal of Allergy and Clinical Immunology. 133 (2), 468 (2014).
- Jones, S. M., et al. Epicutaneous immunotherapy for the treatment of peanut allergy in children and young adults. Journal of Allergy and Clinical Immunology. 139 (4), 1242 (2017).
- Varshney, P., et al. A randomized controlled study of peanut oral immunotherapy: clinical desensitization and modulation of the allergic response. Journal of Allergy Clinical Immunology. 127 (3), 654-660 (2011).
- Anagnostou, K., et al. Assessing the efficacy of oral immunotherapy for the desensitisation of peanut allergy in children (STOP II): a phase 2 randomised controlled trial. Lancet. 383 (9925), 1297-1304 (2014).
- Sampson, H. A., et al. Effect of Varying Doses of Epicutaneous Immunotherapy vs Placebo on Reaction to Peanut Protein Exposure Among Patients With Peanut Sensitivity: A Randomized Clinical Trial. The Journal of the American Medical Association. 318 (18), 1798-1809 (2017).
- Bednar, K. J., et al. Human CD22 Inhibits Murine B Cell Receptor Activation in a Human CD22 Transgenic Mouse Model. Journal Immunology. 199 (9), 3116-3128 (2017).
- Macauley, M. S., et al. Antigenic liposomes displaying CD22 ligands induce antigen-specific B cell apoptosis. Journal Clinical Investigation. 123 (7), 3074-3083 (2013).
- Orgel, K. A., et al. Exploiting CD22 on antigen-specific B cells to prevent allergy to the major peanut allergen Ara h 2. Journal Allergy Clinical Immunology. 139 (1), 366-369 (2017).
- Smarr, C. B., Hsu, C. L., Byrne, A. J., Miller, S. D., Bryce, P. J. Antigen-fixed leukocytes tolerize Th2 responses in mouse models of allergy. Journal of Immunology. 187 (10), 5090-5098 (2011).
- Kulis, M., et al. The 2S albumin allergens of Arachis hypogaea, Ara h 2 and Ara h 6, are the major elicitors of anaphylaxis and can effectively desensitize peanut-allergic mice. Clinical and Experimental Allergy. 42 (2), 326-336 (2012).
- Dang, T. D., et al. Increasing the accuracy of peanut allergy diagnosis by using Ara h 2. Journal of Allergy Clinical Immunology. 129 (4), 1056-1063 (2012).
- Loughrey, H. C., Choi, L. S., Cullis, P. R., Bally, M. B. Optimized procedures for the coupling of proteins to liposomes. Journal Immunological Methods. 132 (1), 25-35 (1990).
- Sen, M., et al. Protein structure plays a critical role in peanut allergen stability and may determine immunodominant IgE-binding epitopes. Journal of Immunology. 169 (2), 882-887 (2002).
- Krall, N., da Cruz, F. P., Boutureira, O., Bernardes, G. J. Site-selective protein-modification chemistry for basic biology and drug development. Nature Chemistry. 8 (2), 103-113 (2016).
- Jimenez-Saiz, R., et al. Lifelong memory responses perpetuate humoral TH2 immunity and anaphylaxis in food allergy. Journal Allergy and Clinical Immunology. 140 (6), 1604-1615 (2017).
- Moutsoglou, D. M., Dreskin, S. C. Prolonged Treatment of Peanut-Allergic Mice with Bortezomib Significantly Reduces Serum Anti-Peanut IgE but Does Not Affect Allergic Symptoms. International Archives of Allergy and Immunology. 170 (4), 257-261 (2016).
- LaMothe, R. A., et al. Tolerogenic Nanoparticles Induce Antigen-Specific Regulatory T Cells and Provide Therapeutic Efficacy and Transferrable Tolerance against Experimental Autoimmune Encephalomyelitis. Frontiers in Immunology. 9, 281 (2018).
- Srivastava, K. D., et al. Investigation of peanut oral immunotherapy with CpG/peanut nanoparticles in a murine model of peanut allergy. J Allergy Clin Immunol. 138 (2), 536-543 (2016).
- Bellinghausen, I., Saloga, J. Analysis of allergic immune responses in humanized mice. Cellular Immunology. 308, 7-12 (2016).
- Pillai, S., Mattoo, H., Cariappa, A. B cells and autoimmunity. Curr Opin Immunol. 23 (6), 721-731 (2011).
- Mantegazza, R., Cordiglieri, C., Consonni, A., Baggi, F. Animal models of myasthenia gravis: utility and limitations. International Journal of General Medicine. 9, 53-64 (2016).
- Berman, P. W., Patrick, J. Experimental myasthenia gravis. A murine system. J Exp Med. 151 (1), 204-223 (1980).
- Berman, P. W., Patrick, J. Linkage between the frequency of muscular weakness and loci that regulate immune responsiveness in murine experimental myasthenia gravis. J Exp Med. 152 (3), 507-520 (1980).
- Derksen, V., Huizinga, T. W. J., van der Woude, D. The role of autoantibodies in the pathophysiology of rheumatoid arthritis. Seminars in Immunopathology. 39 (4), 437-446 (2017).
