After myo6b:GCaMP6s-caax transgenic fish are properly immobilized and the fluid-jet stimulus is delivered to lateral-line hair cells, robust calcium signals can be visualized and measured (Figures 4 and 5, taken at 2 X binning). During fluid-jet stimulation, calcium signals can either be measured in the apical hair bundles, where MET channels open in response to stimuli, or at the base of hair cells, where presynaptic Cav1.3 calcium channels trigger neurotransmission. A representative example of calcium responses in these regions with an individual neuromast are shown in Figure 4A1-A2''. In this example, a 2-s 5 Hz fluid-jet stimulus was delivered to activate all hair cells within the representative neuromast. During the stimulus, robust calcium signals can be detected in hair bundles (Figure 4A1-A1'', responses from 8 hair bundles are shown). In this system, nearly all mature hair cells display this apical influx of calcium5. In contrast, within the same neuromast, there are detectable calcium signals in the basal, synaptic plane in only a subset (~30%) of hair cells (Figure 4A2-A2'', 4 cells with presynaptic responses are shown)5. The 4 green ROIs show cells with no significant presynaptic calcium signals (Figure 4A2-A2'') despite robust apical calcium signals (Figure 4A1-A1''). In this representative example (Figure 4A1-A2''), colored ROIs match up hair bundles in the apical MET plane (Figure 4A1) with their cell bodies in the basal synaptic plane (Figure 4A2). This example highlights how both MET dependent- and presynaptic-calcium signals can be measured within individual hair cells and among populations of hair cells.
The calcium signals in both the hair bundles and at the presynapse can be plotted graphically as either raw (F) GCaMP6s intensity or ΔF/Fo GCaMP6s intensity (see step 12, Figures 4A1'-A1'' and 4A2'-A2''). The (F) GaMP6s graphs highlight that the baseline fluorescence intensity for each cell can differ (Figures 4A1' and 4A2'). In the ΔF/Fo GCaMP6s graphs, each cell is normalized to its baseline value and the relative intensity change from baseline is plotted (Figures 4A1'' and 4A2''). In both the (F) and ΔF/Fo GCaMP6s plots, the calcium signals in both the apical hair bundle and basal presynaptic plane initiate with the onset of the stimulus (gray box) and decline exponentially after the stimulus ends. During the stimulus, calcium signals in hair bundles rise rapidly and saturate if the strength of the deflection does not change (Figure 4A1-A1''). In contrast, within the subset of hair cells with detectable calcium signals in the synaptic plane, the calcium signals increase more gradually and are less prone to saturation (Figure 4A2-A2''). In hair cells without presynaptic calcium signals (green ROIs), the calcium signals remain near baseline.
In addition to these graphical representations (Figure 4), calcium signals can be visualized spatially within the entire neuromast during the time course of the recording. An example of a spatiotemporal representation is shown in Figure 5 for presynaptic GCaMP6s signals in the basal plane of a neuromast. In Figure 5, the main steps to process an image sequence for spatial visualization are outlined as described in step 13. First, the raw (F) GCaMP6s images are temporally binned [Figure 5: row 1 (5 of the 14 bins are shown); step 13.2.1]. Then, the baseline image, calculated from pre-stimulus frames (step 13.2), is subtracted from the (F) GCaMP6s fluorescence signals to obtain ΔF images (Figure 5: row 2; step 13.2.2). Next, the ΔF grayscale images are converted to a color LUT (Figure 5: row 3, Red Hot LUT; step 13.3). Finally, the ΔF images with the LUT conversion are overlaid onto the temporally binned (F) images (Figure 5, first row) to reveal the spatiotemporal signals within the neuromast during stimulation (Figure 5: row 4; step 13.4). The heat maps of ΔF GCaMP6s signals provide both valuable spatial and temporal information that is not easy to parse out from single ROIs and the graphs used in Figure 4. Heat maps can help visualize critical spatiotemporal information, including subcellular information regarding the onset and duration of calcium signals within each hair cell as well as the timing and intensity differences among hair cells within the entire neuromast.
It is important to verify that the graphs and spatial heat maps represent true calcium signals and are not artifacts due to motion. In this protocol, motion artifacts can be the result of excessive drift or movement of the larva or motion due to fluid-jet stimulation. All of these artifacts are challenging to completely eliminate in this in vivo preparation. While registration of image sequences (step 12.1.3) can correct for the majority of movement in x- and y-axes, image sequences with excessive movement in the z-axis must be identified and removed from analyses. Motion artifacts are easiest to identify by graphing the calcium signals. Examples of GCaMP6s intensity changes that are artifacts and are not true GCaMP6s signals can be observed at the apex (Figure 4B1'-B1'') and base (Figure 4C1'-C1'') of hair cells.
In the apical hair bundles, motion artifacts are common when the fluid-jet stimulus is too strong (Figure 3A3'''). During these excessively strong stimuli, the apical hair-bundle plane can move out of focus during fluid-jet stimulation then return to the original focal plane after the stimulus terminates (Figure 4B1'-B''). This makes it difficult to accurately measure apical MET-dependent calcium signals. An example of hair-bundle motion artifacts can be seen in Figure 4B1'-B1''. Here, the graphs show a decrease in GCaMP6s signals during the stimulus (gray box) when the hair bundles are out-of-focus. After the stimulus ends, the GCaMP6s signals rapidly increase as the hair bundles return to their original position and come back into focus. This contrasts with the example in Figure 4A1'-A1'' in which the apical calcium signals increase at the onset of the stimulus and decrease when the stimulus ends.
While movement due to excessive fluid-jet stimuli can also move the synaptic plane out of focus, this type of motion artifact is less common in this plane. Instead, changes in focus in the Z-axis due to movement or drift of the larva are the most common causes of motion artifacts. Larval motion or drift can affect GCaMP6s measurements at both the apex and base of hair cells. An example of larval motion that increases GCaMP6s in the basal, synaptic plane during the stimulus is shown in Figure 4C'-C''. Motion artifacts (Figure 4C1'-C1'') can be distinguished from true presynaptic signals (Figure 4A2'-A2'') by examining the time course of the GCaMP6s signals. Rather than increasing and decreasing exponentially with the stimulus (Figure 4A2'-A2''), the motion-induced increases in GCaMP6s signal have a square shape and rise and fall abruptly with the onset and offset of the stimulus, respectively (Figure 4C1'-C1'').
In addition to careful examination of the time course of GCaMP6s signals, control experiments using pharmacology can be used to differentiate true GCaMP6s signals from motion artifacts. For example, BAPTA (step 10) can be applied to cleave the tip-links that are required for MET-channel function in hair bundles. BAPTA should eliminate both fluid-jet-evoked apical MET-channel-dependent calcium influx as well as the subsequent basal, presynaptic calcium influx through Cav1.3 channels. In the representative example of true stimulus-evoked calcium signals (Figure 4A1-A2'') during fluid-jet stimulation, all changes in GCaMP6s fluorescence in both the apical and basal planes would be eliminated after BAPTA treatment. In contrast, changes in GCaMP6s fluorescence due to motion such as those shown in Figures 4B1'-B1'' and 4C1'-C1'' would not be eliminated by BAPTA treatment.
In addition to using BAPTA to eliminate all stimulus-evoked GCaMP6s signals, isradipine can be applied (step 11) to specifically block Cav1.3-dependent calcium influx in the basal synaptic plane while leaving apical MET-channel-dependent calcium influx intact5. After application of isradipine, in an individual neuromast with no motion artifacts, changes in GCaMP6s fluorescence in apical hair bundles (Figure 4A1'-A1'') during fluid-jet stimulation would be unaltered, while all synaptic GCaMP6s fluorescence changes at the base would be eliminated (Figure 4A2'-A2''). Any change in GCaMP6s signal in the synaptic plane after isradipine application (e.g.,Figure 4C1-C1'') would most likely correspond to motion artifacts.
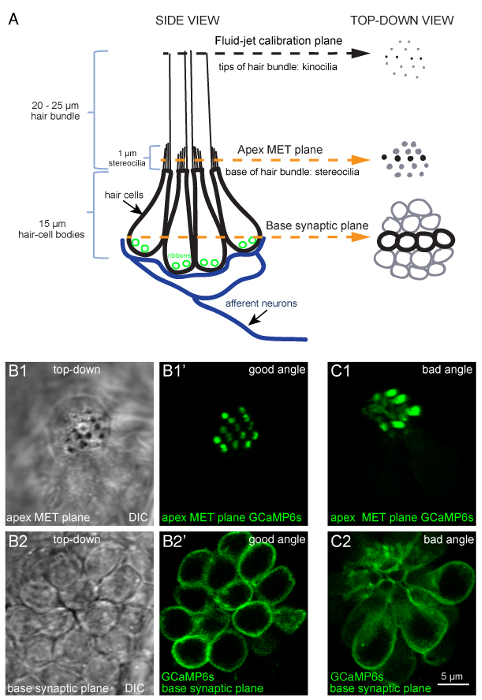
Figure 1: Overview of a lateral-line neuromast and functional imaging planes. (A) The diagram to the left depicts a side-view of a neuromast with four hair-cell bodies (black) contacting postsynaptic afferent neurons (blue). Ribbons (green) tether vesicles at presynaptic active sites within each cell. Apical to each cell body is a bundle of stereocilia (1 µm) that contain MET channels. Each hair bundle has one kinocilium that transfers the mechanical force of water motion to the base of the hair bundle. The diagram on the right depicts the same model in a top-down view. In this top-down view, black is used to indicate the four cells depicted in the diagram on the left, and gray is used to indicate other cells in the neuromast. Within this model and these 2 views, three important planes are highlighted: (1) the tips of the hair bundles (kinocilia) used to quantify the magnitude of hair-bundle deflection, (2) the apical MET plane at the base of the hair bundles where calcium enters the cell during stimulation, and (3) the synaptic plane at the base of the cell where calcium enters near synaptic ribbons. (B1-B1') DIC and GCaMP6s top-down images of MET plane at the base of the hair bundles, where mechanosensation-dependent calcium signals can be recorded. (B2-B2') DIC and GCaMP6s top-down images from the same neuromast as B1-B1', but at the base of the neuromast in the synaptic plane, where presynaptic calcium signals can be detected. (C1-C2) Images of a neuromast expressing GCaMP6s where the larvae is improperly mounted. In this example, the apical MET plane (C1) and synaptic plane (C2) at the base of the cell are positioned at a suboptimal angle. This position does not allow for all hair bundles to be imaged in a single plane, and many more imaging planes are needed to capture activity at all synapses within this neuromast compared to B1-B2'. Images are of larvae at 5 dpf. The scale bar in C2 corresponds to all images in B1-C2. Please click here to view a larger version of this figure.
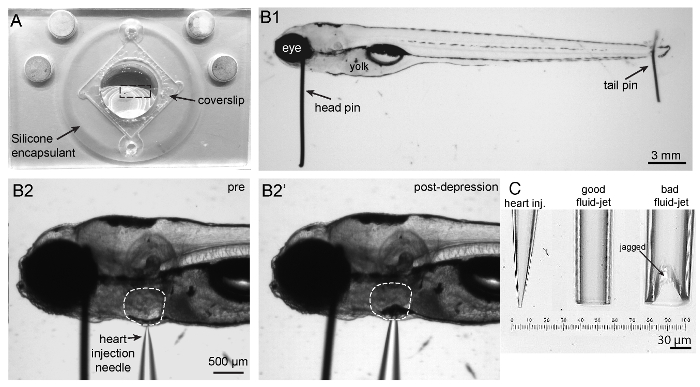
Figure 2: Imaging chamber, zebrafish mounting and heart injection procedures, and needles. (A) Shown is an imaging chamber with a larva (outlined by a dashed rectangle) pinned to the center atop the silicone encapsulant. (B1) Shown is a 5 dpf larva immobilized by two pins. A large head pin is placed perpendicular to the body just posterior to the eye. The two eyes are completely superimposed so the bottom eye is entirely obscured by the upper eye. A small tail pin intersects the notochord in the tail. The larva is flat and not twisted. (B2) To paralyze larva, a heart injection needle is oriented perpendicular to the body and brought adjacent to the heart. The heart injection needle should contact the pigment cell in front of the heart. (B2') Depression of the needle into the skin causes indentation of the pigment cell in front of the heart. (C) Needles in order from left to right: example of a heart injection needle with an opening of approximately 3 µm; example of a good fluid-jet needle with an opening of approximately 50 µm; example of a poorly broken fluid-jet needle that is large and jagged and will likely produce excessive and irregular stimuli. Please click here to view a larger version of this figure.
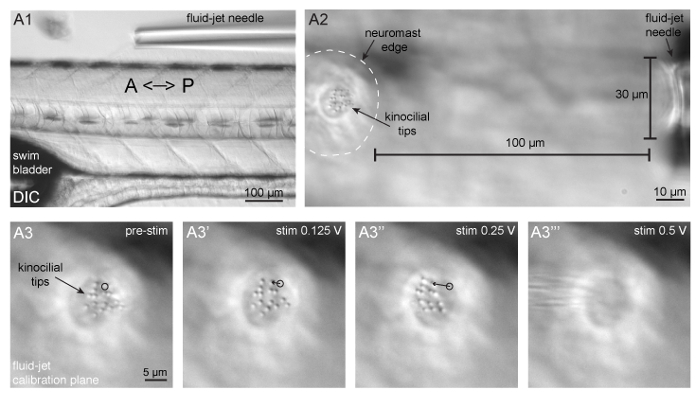
Figure 3: Fluid-jet alignment, positioning, and stimulus calibration. (A1) Shown is a larva oriented with the head facing to the left and tail to the right, and a fluid-jet needle oriented parallel to the A-P axis of the zebrafish body. This fluid-jet needle is aligned to stimulate the neuromasts that respond to anterior (push/pressure) and posterior (pull/vacuum) directed fluid-flow. (A2) Shown is a neuromast (outlined by dashed white line) and tips of apical hair bundles (kinocilia) on the left side the panel and the fluid-jet needle on the right side of the panel. The fluid-jet is positioned approximately 100 µm from the edge of the neuromast. (A3-A3''') The tips of apical hair bundles (kinocilia) are deflected different distances by varying fluid-jet stimulus pressures. The trajectory of a single kinocilial tip is shown for 1.5 µm (A3') and 5 µm (A3'') deflection distances. The black circle indicates the resting position of the kinocilium. It is important that kinocilia are not deflected too far, otherwise the stimulus intensity cannot be reliably quantified and can become damaging (A3'''). Please click here to view a larger version of this figure.
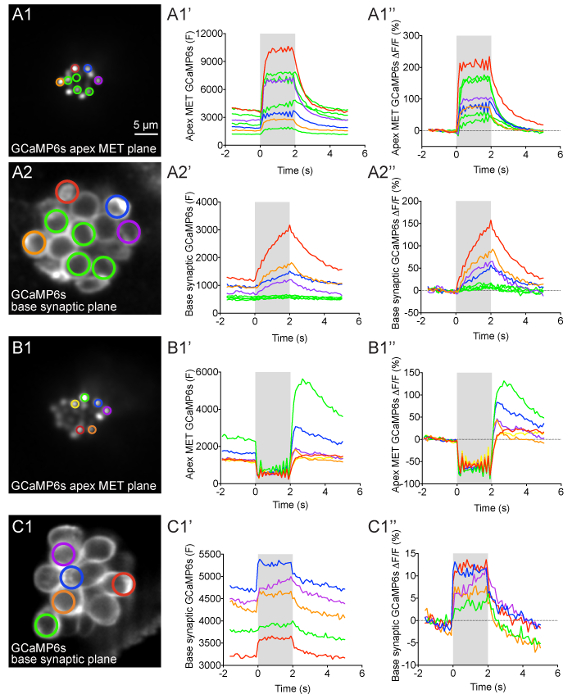
Figure 4: Apical MET and basal presynaptic GCaMP6s signals during fluid-jet stimulation in lateral-line hair cells. (A1-A2'') GCaMP6s intensity changes during fluid-jet stimulation within a representative neuromast. The images on the left show the apical MET plane (A1) and basal synaptic plane (A2) within the same neuromast. The ROIs color coded in A1 and A2 were used to plot the time course of (F) and ΔF/F GCaMP6s intensity graphs to the right of each image. (B1-B1'') Example of an apical MET image sequence with excess movement during fluid-jet stimulation. The image on the left (B1) shows the ROIs used to plot the (F) and ΔF/F GCaMP6s intensity graphs to the right. (C1-C1'') Example of an image sequence in the basal synaptic plane that shows movement artifacts and GCaMP6s signal changes that are not true calcium signals. The image on the left (C1) shows the ROIs used to plot the (F) and ΔF/F GCaMP6s intensity graphs to the right. The gray box in each graph represents the duration of the fluid-jet stimulus during each image sequence. A 2-s 5 Hz fluid-jet stimulus was used for the example in A1-A2'' and B1-B1''. In C1-C1'', a 2 s anterior step stimulus was used. The Y axis for (F) GCaMP6s graphs depicts arbitrary units (A.U.) obtained from Fiji image intensity measurements. All examples are from larvae at 4-5 dpf. Scale bar = 5 µm for all images. Please click here to view a larger version of this figure.
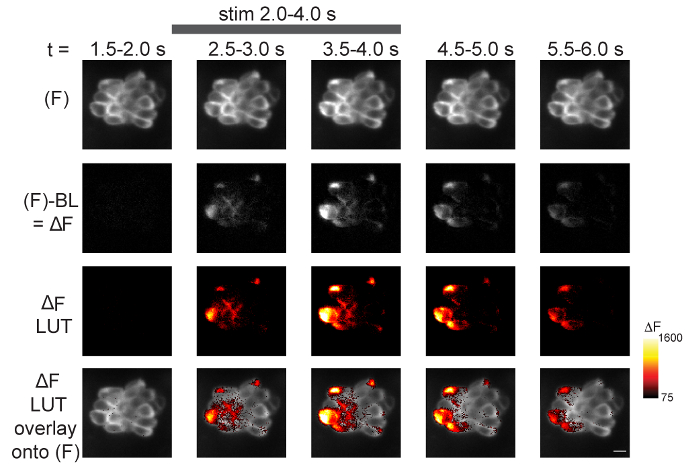
Figure 5: Spatiotemporal visualization of presynaptic GCaMP6s signals during fluid-jet stimulation. The steps to visualize the spatiotemporal changes in GCaMP6s intensity within a neuromast during stimulus are outlined. Time is represented from left to right according to the time stamp at the top of the images. The top row shows 5 of the 14 temporal bins from a 70-frame GCaMP6s (F) image sequence (step 13.2.1). In the second row, the baseline (step 13.2) has been removed from each (F) GCaMP6s binned image to create ΔF images (step 13.2.2). In the third row, ΔF images have been converted from grayscale (second row) to Red Hot LUT (step 13.3). The min and max of these LUT images are set according to the Red Hot LUT heat map of relative ΔF intensity (A.U.) on the right (step 13.3.1). In the bottom row, the third row has been overlaid onto the (F) images in the top row (step 13.4). The gray bar at the top of the figure indicates the timing of the 2-s 5 Hz fluid-jet stimulus. The example is from a 5 dpf larvae. A legend of the Red Hot LUT heat map of relative ΔF intensity (A.U.) is shown to the right. Scale bar = 5 µm for all images. Please click here to view a larger version of this figure.
Supplemental Coding File. Please click here to download this file.
