mRNA electroporation is more efficient than DNA electroporation
We used pCS2+.H2B-Citrine to prepare in vitro transcribed mRNA. Since DNA electroporation is usually performed at 1-2 µg/µL, we used an equimolar concentration of mRNA (calculated to be around 0.25-0.5 µg/µL for H2B-Citrine) for mRNA electroporation. We first tested the electroporation efficiency of pCS2+.H2B-Citrine DNA compared to H2B-Citrine mRNA (in vitro transcribed from the SP6 promoter of pCS2+.H2B-Citrine) by electroporating DNA or mRNA separately into HH5 quail embryos then examining electroporation efficiency at 12 h post-electroporation. Although DNA electroporation leads to brighter fluorescence in some electroporated cells, the efficiency of DNA electroporation was visibly lower compared to the widely-expressed mRNA encoded FPs (Figure 2A and B).
To account for inherent embryo-to-embryo variability in the electroporation protocol, an equimolar combination of mRNA encoding H2B-Citrine and DNA expressing H2B-mKate2 was electroporated into the ectoderm of HH5 embryos and observed 12 h post-electroporation. When comparing DNA and mRNA co-electroporation in the same embryo, DNA expressing FP was also significantly and consistently less efficient than that of mRNA (Figure 2C). By quantifying all embryos electroporated with only mRNA, DNA, or a combination of the two, we found that mRNA transfects ~75% of cells in a given region, while DNA only transfects ~25% of cells in a given region (Figure 2D). The spread in the expression of fluorescent proteins encoded by mRNA or DNA was not significantly different in all co-electroporated embryos (Figure 2E).
Protein expression is quicker in mRNA electroporation than DNA electroporation
The transfected mRNA should lead to faster protein production since it can be immediately recognized by cytosolic translation machinery as seen in Figure 3. DNA must be translocated to the nucleus, transcribed, and the mRNA transported back to the cytosol where it is recognized by cytosolic translation machinery, expected to take more time. To directly compare mRNA versus DNA expression rates of protein production, we carried out a series of time course experiments in which we co-electroporated an equimolar combination of DNA (pCMV.H2B-mKate2) and mRNA (H2B-Citrine) into the ectoderm of HH5 embryos. We detected mRNA-encoded FP expression around 22 min post-electroporation, which continues increasing in relative fluorescence intensity for ~6 h (Figure 3A-E, Supplemental. Movie S1b). In contrast, DNA-encoded FP expression is first seen at ~1.5 h post-electroporation (Figure 3A’-E’, Supplemental. Movie S1c) and continues increasing in brightness until 6 h when the movie was stopped. Because the DNA electroporated cells were saturated by the 6 h mark, we reimaged this condition with weaker imaging conditions (Figure 3F-F’’). Across all time points of this time lapse (22 min to 6 h), mRNA transfects several times more cells than DNA (Supplemental. Movie S1a, the total efficiency of electroporation through brightfield and DAPI are not shown because these images are taken from a time-lapse of a wild type quail embryo). Based on this, we conclude that mRNA electroporation leads to earlier expressed protein.
Co-electroporation of multiple mRNAs is highly efficient
Next, we sought to further test the efficiency of mRNA electroporation by electroporating multiple mRNAs into a region of interest. Co-electroporation of multiple DNAs had previously been shown to be relatively inefficient, showing with the number of multi-labeled being 25%, 14%, and 10% for 2, 3, and 4 DNA constructs electroporated, calculated as a proportion of the total number of cells11. We first electroporated two mRNAs that encode spectrally distinct FPs (Turquoise2-Golgi/H2B-Citrine) into HH5 embryos and obtained high transfection efficiency in all electroporated regions. We used this double-electroporation to capture movies of radial expansion between area pellucida and opaca in a HH4 gastrulating embryo, imaged 2 h post-electroporation (Supplemental Movie S2a, b, and c). The H2B-Citrine is localized within the cell nucleus and permits cell proliferation to be traced27. The Turquoise2-Golgi FP appears to show subcellular polarity within the embryonic cells but does not appear to correlate with speed or direction of moving ectoderm cells in vivo28. This movie (Supplemental Movie S2) shows that cells electroporated with multiple mRNAs can continue normal cellular activity without any apparent defects.
Furthermore, co-electroporation of 4 mRNAs – Turquoise2-Golgi (to visualize Golgi apparatus), LifeAct-eGFP (to visualize F-actin), H2B-Citrine (to visualize nucleus), and membrane-Cherry FP (to visualize membranes) — resulted in 87% transfection efficiency of all 4 mRNAs in all electroporated regions (Figure 4). LifeAct-EGFP and H2B-Citrine were separated by the linear unmixing processing tool in the commercial software.
FRAP assay shows the rapid degradation of electroporated mRNA during the first two hours post-electroporation.
It is well-known that naked mRNAs are quickly degraded by cellular RNAases29. We postulated that mRNA electroporation might be useful for loss or gain-of-function experiments by allowing fast and efficient expression of proteins in a developing embryo. Therefore, it would be helpful to quantitate the rate of mRNA decay after mRNA electroporation, since mRNA degrades faster than DNA in cells. In previous reports of mRNA electroporation in adult mouse neural stem cells, around 80% mRNA was identified 2 h post-electroporation by qRT-PCR, yet little to no mRNA was detected by 24 h post-electroporation15. We sought to further quantify mRNA expression post-electroporation by designing an in vivo FRAP assay to detect mRNA levels in electroporated cells.
Photobleaching is the irreversible destruction of the fluorophore that can occur when the fluorophore (fluorescent proteins in our case) is in an excited state, which eliminates fluorescence during observation30,31. Any emitted light from fluorescent proteins (FPs) that can be detected in photobleached cells above baseline levels should, therefore, represent newly translated FPs encoded by intact, transfected mRNAs. Therefore, we sought to photobleach the electroporated cells to eliminate all existing FP-derived fluorescence at a variety of time points and track subsequent fluorescence recovery.
Using optimized photobleaching settings as described in the protocol section, we found that photobleaching initially reduces the fluorescence intensity in all photobleached cells by ~95% when assayed immediately after the photobleaching event. This was done at 45 min, 2 h, and 5 h post-electroporation (Figure 5A-C). The fluorescence recovery in each cell within the photobleached region was tracked over a 30 min time period after the photobleaching at each time point by imaging the same region at regular time intervals (3 or 5 min). Our data suggests that there is a significant decrease in transfected mRNA levels between 45 min and 5 h post-electroporation. This is shown by the large decrease in FRAP measured at 5 h compared to 45 min post-electroporation. To highlight the fluorescence recovery in the bleached cells especially in the 5 h time point, we enhanced the brightness in Figure 5A-C using the brightness/contrast tool in ImageJ and show these modified images in Figure 5D-F. Interestingly, there is a large heterogeneity in fluorescence recovery from cell-to-cell within the photobleached area at 2 h because some cells recover fluorescence faster than other cells (Figure 5E’’, see arrowheads). However, all cells within the 5 h photobleached area recover fluorescence slower (Figure 5F’’) than the photobleached cells at 2 h. We also detect actively dividing cells within our photobleached region, suggesting that cells have not died as a result of the photobleaching process (Figure 5E’’).
We quantify these results in Figure 6A by showing the normalized signal intensity of each cell within the photobleached areas over a 30 min period after the photobleaching event. For each cell, the fluorescence values were normalized against the signal intensity of that cell immediately after photobleaching. This normalization was done for all cells across all time points imaged (45 min, 2 h, 5 h). The normalized values of fluorescence recovery of the cells within the same photo bleached region were then pooled together and graphed over time after photobleaching event; therefore, each point in the graph represents ~35 cells. The unnormalized data is also shown in Figure 6B, in which each line represents a cell with recovering fluorescence signal intensity immediately after photobleaching. This data overall suggests that by 5 h, all cells have depleted most of their mRNA, with a large portion of this drop occurring between 45 min and 2 h post-electroporation.
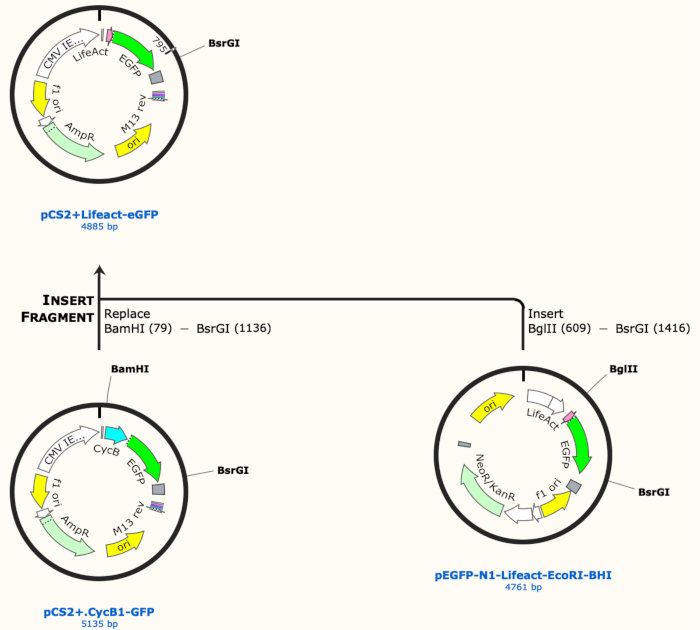
Figure 1: Schematic of cloning procedure to make pCS2+LifeAct-eGFP construct. Please click here to view a larger version of this figure.
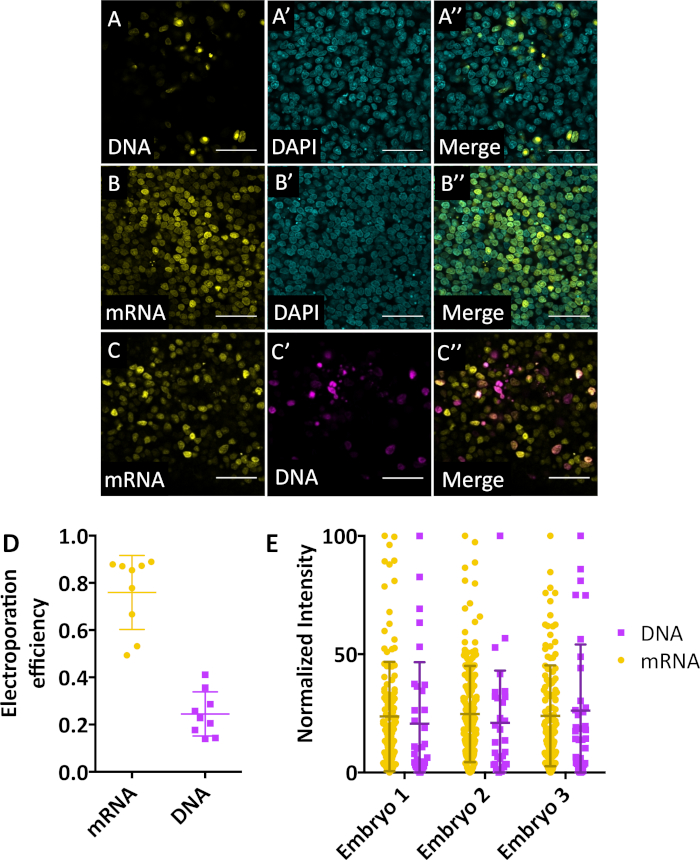
Figure 2: mRNA electroporation is more efficient than DNA electroporation. (A-A’’) DNA expressing pCS2.H2B-Citrine was electroporated into the ectoderm of HH5 embryos. Fluorescence was observed 12 h post-electroporation. Scale bar = 50 µm. (B-B’’) mRNA expressing H2B-Citrine was electroporated into the ectoderm of HH5 embryos. Fluorescence was observed 12 hours post-electroporation. Scale bar = 50 µm. (C-C’’) Equimolar combination of DNA (pCMV.H2B-mKate2) and mRNA (H2B-Citrine) was electroporated into HH5 embryos and observed 12 h post-electroporation. Three embryos were tested per condition (n = 2 experiments). Scale bar = 50 µm. (D) Three embryos from A, B, and C (9 total embryos) were quantified for electroporation efficiency using particle analysis tool on ImageJ. A 200 µm region from each embryo was quantified (at least 150 cells were counted per region). Middle dash represents mean, while top and bottom bars represent standard deviation from the mean. (E) Three co-electroporated (DNA and mRNA) embryos are analyzed for spread of signal across all electroporated cells in a given 200 µm region. Each embryo had around 100 cells positive for mRNA electroporation and 30 cells positive for DNA electroporation. Middle dash represents mean, while top and bottom bars represent standard deviation from the mean. There is no significant difference between the spread of mRNA vs. DNA electroporation, but mRNA electroporation is much more efficient than that of DNA. (A-C) Dimensions = 425.10 x 425.10 x 55 µm. Pixel dwell 2.55 µs. Average line 4. Bits per pixel = 16. Microscope metadata = inverted confocal microscope, 20x objective (see Table of Materials) (A) Ex: 488 nm (0.05%), Emission Track S1 = 508-579 nm; (B) Ex = 488 nm (5.0%), Emission Track S1 = 508-579 nm; (C) Ex = 488 nm (5.0%), 561 nm (5.0%), Emission Track S1 = 508-579 nm; S2 = 606-695 nm Please click here to view a larger version of this figure.
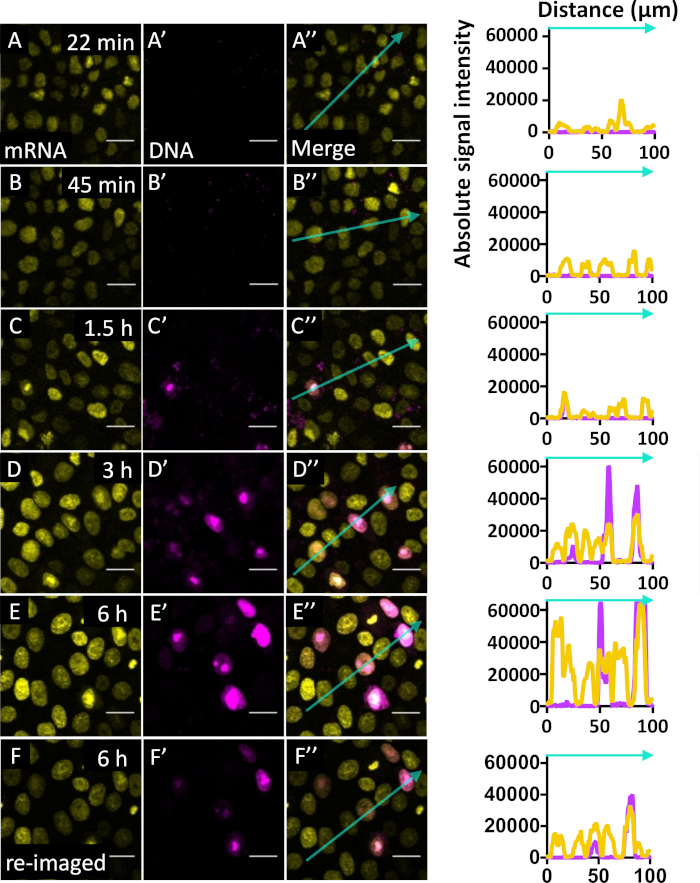
Figure 3: mRNA electroporation shows expression by 22 min post-electroporation and is much quicker than DNA electroporation. Representative images of time course after mRNA (H2B-Citrine) and DNA (pCMV.H2B-mKate2) electroporation with plot profile analysis at 22 min (A-A’’), 45 min (B-B’’), 1.5 h (C-C’’), 3 h (D-D’’), and 6 h (E-E’’) at the start of imaging (n = 1). Plot profile analysis is provided for each time point shown across the drawn arrow in the merged image. Each peak in the plot profile represents the nucleus of an electroporated cell. The cells generally become brighter over the 6 h interval of the movie, indicating that mRNA is still being translated into new fluorescent protein. For the 6 hour time point, images of the same region captured using identical imaging conditions (E-E’’) and weaker imaging conditions (F-F’’) are shown because the images of the DNA electroporated cells were highly saturated using the initial imaging conditions. Scale bar = 20 µm. Dimensions: 425.10 x 425.10 x 55 µm. Image is shown as a maximum intensity projection across all time points imaged. Pixel dwell 2.55 µs. Average line 4. Bits per pixel = 16. Microscope metadata = inverted confocal microscope, 20x objective (see Table of Materials) Ex = 488 nm (15%), 594 nm (5.0%) for initial; Ex = 488 nm (7.5%), 594 nm (0.5%) for last time point (Figure 2F), Emission Track S1 = 499-562 nm; S2 = 605-661 nm. Please click here to view a larger version of this figure.
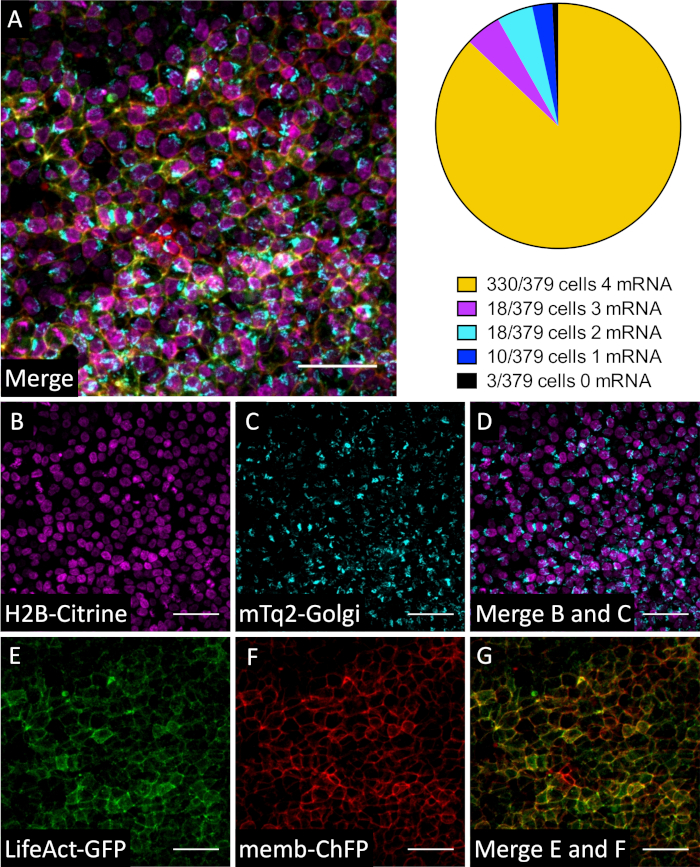
Figure 4: Co-electroporation of four mRNAs targeting different sub-cellular structures is highly efficient. The ectoderm of a HH3 embryo was electroporated with mRNAs that encode mTurquoise2-Golgi, LifeAct-eGFP, H2B-Citrine, and membrane-Cherry. The embryo was fixed and imaged at 4 hours post-electroporation. Most cells are electroporated within the electrode targeted region with all four mRNAs (87%). Representative figures shown (3 embryos each, n = 2 experiments). Scale bar = 50 µm. (A) Merge all four mRNAs (mTurquoise2-Golgi, LifeAct-eGFP, H2B-Citrine, and membrane-Cherry). (B) Only H2B-Citrine channel (Ex = 488 nm (0.7%), Em = 455-499 nm). (C) Only mTurquoise2-Golgi channel (Ex = 458 nm (22.0%), Em = 455-499 nm). (D) Merge H2B-Citrine and mTurquoise2-Golgi shows that Golgi envelopes one side of the nucleus in most cells. (E) Only LifeAct-eGFP channel (Ex = 488 nm (0.7%), Em: 455-499 nm). (F) Only Membrane-Cherry channel (Ex = 594 nm (37.1%), Em = S2: 570-695 nm). (G) Merge LifeAct-eGFP and membrane-Cherry shows overlap in most cells. Dimensions = 425.10 x 425.10. Pixel dwell 1.58 µs. Average line 4. Bits per pixel = 16. Microscope metadata = inverted confocal microscope, 20x objective (see Table of Materials) Ex = 405 nm (2.4%), 458 nm (22.0%), 488 nm (0.7%), 594 nm (37.1%), Emission Track S1 = 455-499 nm; S2 = 570-695 nm. Please click here to view a larger version of this figure.
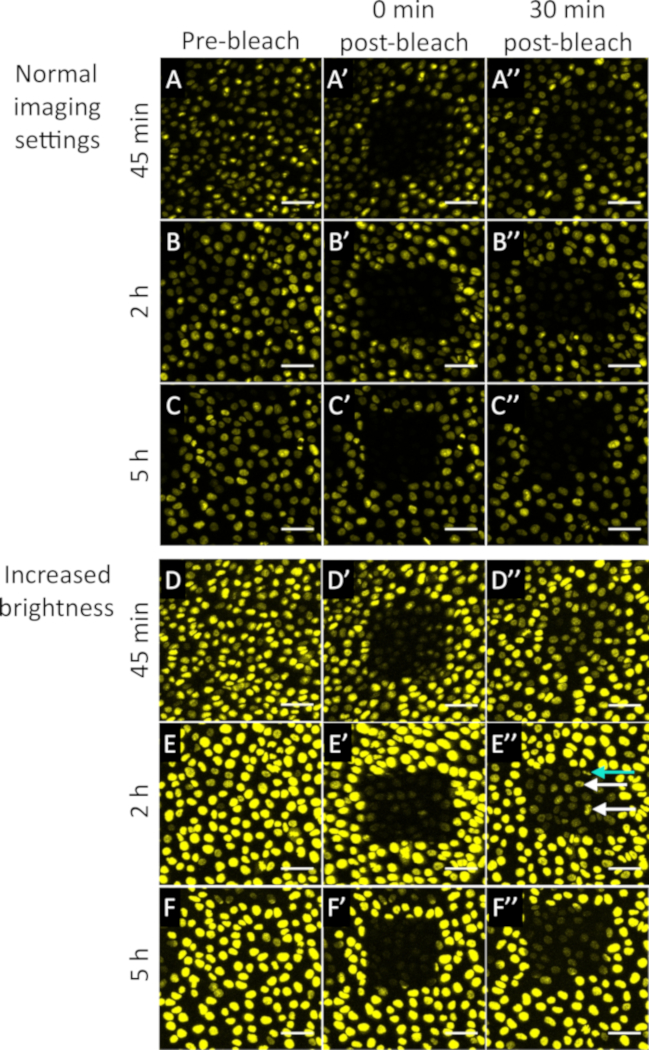
Figure 5: Photobleaching assay shows rapid degradation of electroporated mRNA during the first five hours post-electroporation. Photobleaching of a 100 µm² region containing cells electroporated with mRNA expressing H2B-Citrine is performed at 45 min, 2 h, and 5 h post-electroporation. The embryo was imaged at regular time intervals after the photobleaching (3 or 5 min). Photobleaching conditions are as follows: 70% 405 nm laser, 100 iterations, about 5 min duration for entire region. Scale bar 50 = µm. (A-A’’) 45 min pre- and post-bleach. B shows embryo immediately after photobleaching and C shows embryo 30 min post-bleach. Scale bar = 50 µm. (B-B’') 2 h pre- and post-bleach. The perimeter of the square is shifted because the cells within the photobleached region moved slightly during the bleach period. (C-C’’) 5 h pre- and post-bleach. (D-D’’) Same images as A-A’’ with enhanced brightness (maximum value adjusted to 11550 to enhance brightness post-collection). (E-E’’) Same images as B-B’’ with enhanced brightness (maximum value adjusted to 11550 to enhance brightness post-collection). The white arrowheads point to cells that have higher fluorescence recovery than surrounding cells. The cyan arrowhead points to a photobleached cell that has recently undergone mitosis. (F-F’’) Same images as C-C’’ with enhanced brightness (maximum value adjusted to 11550 to enhance brightness post-collection). Dimensions = 425.10 x 425.10 x 42 µm. Image is shown as a maximum intensity projection across all time points imaged. Pixel dwell 1.58 µs. Average line 4. Bits per pixel = 16. Microscope metadata = inverted confocal microscope, 20x objective (see Table of Materials) Ex = 488nm (1.5%), Emission Track S1 = 508-553 nm Please click here to view a larger version of this figure.
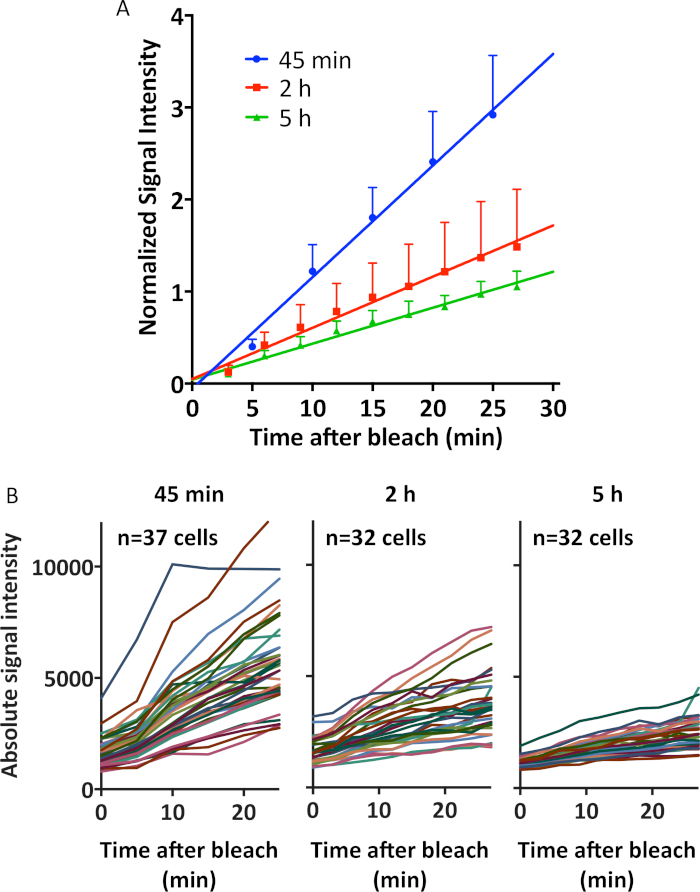
Figure 6: Quantification of the fluorescence recovery in photobleached cells. (A) The fluorescence intensity of all cells within the bleached regions (~35 cells) were tracked for each single cell during each time period post-bleach. There is a large drop in rate of recovered signal intensity from 45 min to 2 h, followed by a smaller drop from 2 h to 5 h. The top error bar is shown for each point, which represents standard deviation from the mean. The line shown is a linear regression line. R² for 45 min, 2 h, and 5 h is 0.83, 0.58, and 0.84 respectively. The top half of the error bar is shown for each point. (B) The fluorescence intensity of all cells within the bleached regions (~35 cells) were tracked for each single cell during each time period post-bleach and graphed without normalization. There is a large drop in rate of recovered signal intensity from 45 min to 2 h, followed by a smaller drop from 2 h to 5 h. Please click here to view a larger version of this figure.
Supplementary movie 1: mRNA electroporation leads to earlier protein expression than DNA electroporation. H2B-Citrine mRNA and H2B-mKate2 DNA electroporated into ectoderm of HH5 embryo. Region imaged is at the border of extra-embryonic and embryonic tissue. Scale bar = 50 µm. (a) Both H2B-Citrine and H2B-mKate2 channel shown (Ex = 488 nm (15%), 594 nm (5.0%), Em: 499-562 nm; S2: 605-661 nm). (b) Only mRNA H2B-Citrine channel shown (Ex = 488 nm (15%), Em = 499-562 nm). (c) Only H2B-mKate2 DNA channel shown (Ex = 594 nm (5.0%), Em = 605-661 nm). Dimensions = 850.19 x 850.19 x 110 µm. Image is shown as a maximum intensity projection across all time points imaged. Pixel dwell 2.55 µs. Average line 4. Bits per pixel = 16. Microscope metadata: inverted confocal microscope, 20x objective (see Table of Materials) Ex = 488 nm (15%), 594 nm (5.0%), Emission Track S1 = 499-562 nm; S2 = 605-661 nm. Please click here to download this file.
Supplementary movie 2: Co-electroporation of mRNAs permits distinct cell behaviors to be dynamically studied. Radial expansion of cells between the embryonic and extra-embryonic border are electroporated with mRNA (H2B-Citrine and mTurquoise2-Golgi) and imaged during outward migration. This movie was imaged by confocal microscopy from 2 to 5 h post-electroporation by imaging every 6 min. Scale bar = 50 µm. (a) Both H2B-Citrine and mTurquoise2-Golgi channels shown (Ex = 458 nm (28%), 488 nm (5.0%), Em = 446-526 nm; S2: 508-553 nm). (b) Only H2B-Citrine channel shown (Ex = 488 nm (5.0%), Em = S2: 508-553 nm). (c) Only mTurquoise2-Golgi channel shown (Ex = 458 nm (28%), Em = 446-526 nm). Dimensions = 425.10 x 425.10 x 32.5 µm. Image is shown as a maximum intensity projection across all time points imaged. Pixel dwell 0.79 µs. Average line 4. Bits per pixel = 16. Microscope metadata = inverted confocal microscope, 20x objective (see Table of Materials) Ex = 458 nm (28%), 488 nm (5.0%), Emission Track S1 = 446-526 nm; S2 = 508-553 nm. Please click here to download this file.
