Through BLI assays, we previously established that binding of GrgA to σ28 is dependent on a 28 amino acid middle region (residues 138-165) of GrgA15. Accordingly, compared with N-terminally His-tagged full length GrgA (NH-GrgA), a GrgA deletion construct lacking this region (NH-GrgAΔ138-165) had a decreased association rate and an increased dissociation rate, leading to a 3 million-fold loss of overall affinity (Table 1). Here, we demonstrate that this middle region directly binds σ28 in the absence of the rest of the GrgA protein. In these experiments, the middle region tagged with an N-terminally His-tag (NH-GrgA138-165) was used as the ligand, which was first immobilized to the tip of a Ni-NTA biosensor (Figure 1A). After washing unbound NH-GrgA138-165 off the biosensor, real-time association with the analyte σ28 was recorded following the addition of σ28. Finally, the real-time dissociation was recorded following the wash. Recordings of experiments with three different analyte concentrations starting 30 s prior to ligand binding and ending 2 minutes after the beginning of wash are shown in Figure 1A. To better visualize the ligand-analyte interaction, we remove data prior to the addition of the ligand and reset the baseline to 0 to derive Figure 1B.
Values of kinetic parameters for interaction of the NH-GrgA138-165 fragment with σ28 are presented in Table 1. Compared to the NH-GrgA X σ28 interaction, the NH-GrgA138-165 X σ28 interaction displayed a trending statistically significant 60% reduction in ka, a highly statistically significant 64% increase in kd, and a highly statistically significant 3.5-fold increase in KD. These changes demonstrate that compared to NH-GrgA, NH-GrgA138-165 binds σ28 more slowly, dissociates from σ28 faster, and has a decreased overall affinity with σ28. Therefore, residues 138-165 in GrgA binds σ28 but with reduced affinity compared to full length GrgA.
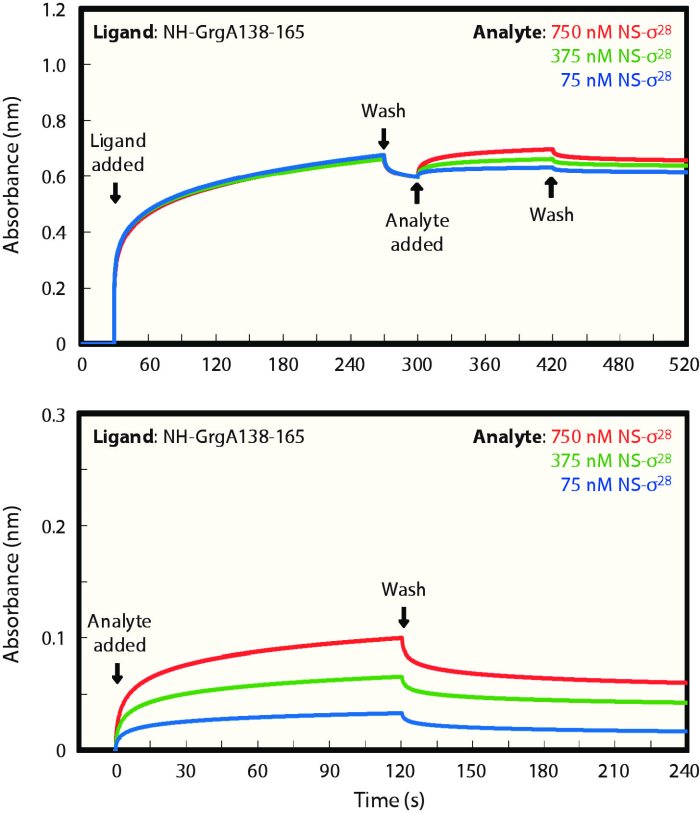
Figure 1: A 28 amino acid middle region of GrgA binds σ28 in vitro.
(A) Real-time changes in light interference patterns recorded by in four stages: (i) binding of NH-GrgA138-165 (Ligand) to a Ni-NTA biosensor, (ii) wash, (iii) binding of NS-σ28 (Analyte) at different concentrations to the immobilized NH-GrgA-138-165 (Ligand), and (iv) subsequent wash. (B) Enhanced visualization of ligand-analyte association and dissociation following removal of values in the first two stages from (A) and reset of the baseline. Panel B is modified from Desai et al., 201815. Please click here to view a larger version of this figure.
| Ligand | n | ka | kd | KD | References | |||||||||||
| 1/Ms | % control | 1/s | % control | M | % control | |||||||||||
| NH-GrgA | 8 | (1.5 ± 1.7) x 104 | 100 | (2.8 ± 0.8) x 10-3 | 100 | (2.2 ± 0.3) x 10-7 | 100 | Desai et al, 2018 | ||||||||
| NH-GrgAΔ138-165 | 2 | (5.6 ± 0.1) x 103 | 37 | (4.1 ± 0.3) x 102 | 1.5 x 107 | (6.9 ± 4.5) x 10-2 | 3.1 x 108 | Desai et al, 2018 | ||||||||
| p=0.125 | p<0.002 | p<0.001 | ||||||||||||||
| NH-GrgA138-165 | 3 | (6.0 ± 1.0) x 103 | 40 | (4.6 ± 0.4) x 10-3 | 164 | (7.7 ± 0.3) x 10-7 | 350 | This study | ||||||||
| p=0.074 | p=0.006 | p<0.001 | ||||||||||||||
Table 1: A mutant of GrgA, containing only amino acid residues 138-165, binds σ28 despite lower affinity compared to the full-length GrgA.
BLI assays were performed with Ni-NTA biosensors using His-tagged full-length GrgA or deletion mutants as ligands and purified Strep-tagged σ28 as an analyte. Graphs of recordings are shown in Figure 1. Values of kinetic parameters (averages ± standard deviations) were generated with the associated software17. ka (association rate constant) is defined as the number of complexes formed per s in a 1 molar solution of A and B. kd (dissociation rate constant) is defined as the number of complexes that decay per second. KD (dissociation equilibrium constant), defined as the concentration at which 50% of ligand binding sites are occupied by the analytes, is kd divided by ka. n, number of experimental repeats. p values were calculated using 2-tailed Student’s t tests. Kinetic parameters for NH-GrgA and NH-GrgAΔ138-165 were from Desai et al., 201815.
