A general scheme of the conditional rescue system
Figure 1 shows the targeting scheme to conditionally rescue EED KO cells with either WT or cage-mutant (Y365A) EED that is expressed from the endogenous EED locus. After knocking out EED, a core subunit of PRC2 that is essential for its stability and enzymatic activity, a cassette within the intron following exon 9 of EED is introduced (Figure 1). The cassette consists of the remaining 3' cDNA sequence of EED, in reverse orientation with respect to the endogenous gene sequence. The cassette is flanked by heterologous inverted loxP sites (lox66 and lox71)18. The cells are propagated until the complete loss of H3K27me2/3 is observed. Upon activation of Cre recombinase expression by the addition of 4-OHT, the cassette is inverted such that exon 9 is spliced into the cassette using the splice acceptor sequence (Figure 1). With this system, EED KO cells can now be rescued by either WT or cage-mutant versions of EED both of which have a C-terminal Flag-HA tag. The downstream T2A-GFP provides a marker to select for cells that undergo a successful inversion event. The polyA signal prevents transcription of downstream sequences. With this system, the kinetics of PRC2 recruitment and the formation of H3K27me domains can now be followed.
Tracking the temporal deposition of PRC2-mediated H3K27me2/3
To follow the temporal dynamics of PRC2-mediated chromatin domain establishment, the WT or cage-mutant version of EED is re-expressed in the background of EED KO cells, upon 4-OHT treatment (Figure 2A,B). To follow deposition of H3K27me2/3 marks, ChIP-seq of H3K27me2/me3 are performed after re-expression of WT or cage-mutant EED for the indicated time points. The emergence of H3K27me3 is observed at 12 h after WT EED expression at discrete regions that are denoted as "nucleation sites" (Figure 2C), and then at regions distant from the initial nucleation sites by 24 h13. Eventually, the distribution of H3K27me3 nearly approximates that of the levels seen in WT parental cells at 36 h after WT EED expression. The temporally established downstream sites of H3K27me3 are termed "spreading sites"13. This system can also track the temporal deposition of H3K27me2, which appears to precede H3K27me3 deposition (Figure 2C,D). Lastly, re-expression of cage-mutant EED exhibits different dynamics relative to WT EED (Figure 2C,D). In this case, deposition of H3K27me3 is very inefficient and instead, H3K27me2 becomes apparent and more concentrated at the nucleation sites, indicating that the cage-mutant EED is unable to spread the modification to neighboring regions.
The emergence of H3K27me3 foci and their growth can also be visualized by microscopy (Figure 2E)13. Before the induction of WT EED expression, H3K27me3 staining is not apparent. However, 12 h of WT EED expression shows evidence of H3K27me3 foci formation. These foci increase in number and size by 24 h, and eventually spread to large regions of the nucleus by 36 h of WT EED expression. This system gives evidence of the de novo formation of PRC2-mediated chromatin domains as monitored by ChIP-seq and immunofluorescence (Figure 2C-E)13.
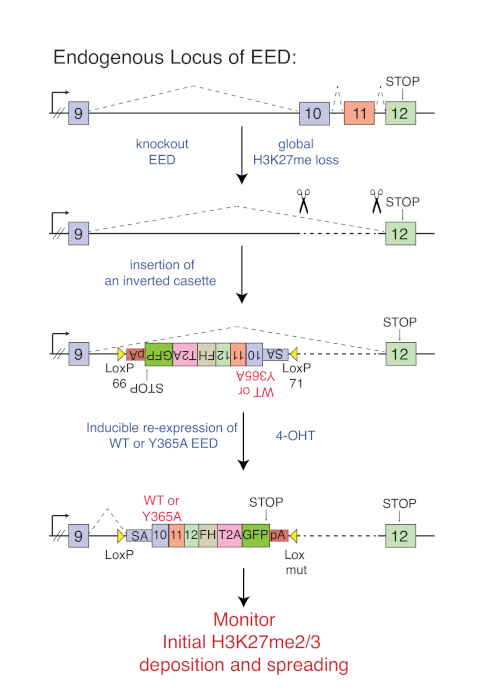
Figure 1: Targeting scheme to conditionally rescue EED KO mESCs either with WT or cage-mt EED (Y365A). Deletion of exon 10 and 11 causes destabilization and degradation of EED and global loss of H3K27me2/me3. A cassette in the intron between exon 9 and 10 is inserted in the reverse direction as indicated. Addition of 4-OHT inverts the cassette such that endogenous exon 9 splices into the cage-mutant or wild type cDNA of the cassette allowing inducible re-expression of WT or cage-mutant EED. This figure has been modified from Oksuz et al., 201813. Please click here to view a larger version of this figure.
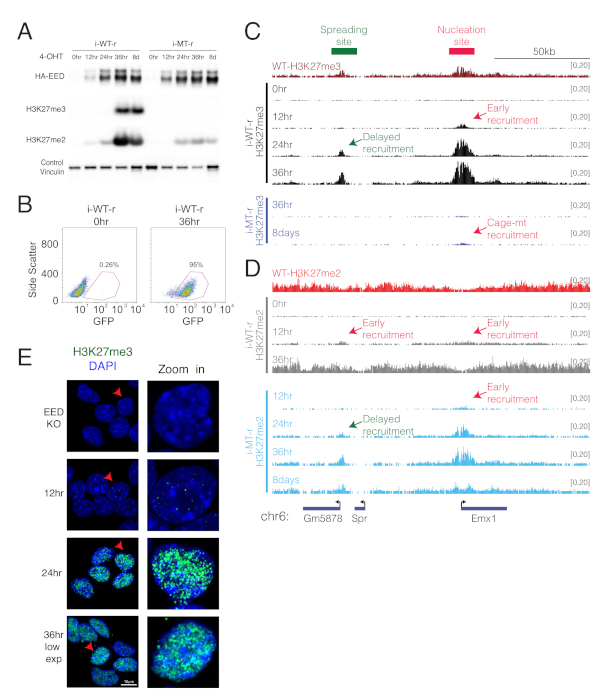
Figure 2: Validations and representative applications of inducible EED rescue systems (i-WT-r and i-MT-r). (A). i-WT-r and i-MT-r systems are validated by Western blot using indicated antibodies on whole extracts after 4-OHT treatment to induce expression of WT (i-WT-r) or cage-mutant (i-MT-r) EED, at the time point indicated. (B). Flow cytometry analysis of GFP in i-WT-r cells before (0 h) and after (36 h) 4-OHT treatment for confirming the efficiency of the expression of T2A-GFP, which is indicative of successful flip of the inverted cassette. (C, D). ChIP-seq tracks for H3K27me3 (C) and H3K27me2 (D) near the Emx1 gene in the WT, or in i-WT-r or in i-MT-r cells, for the indicated times of 4-OHT treatment. Early and delayed sites for deposition of the histone marks are indicated. (E). Immunofluorescence using H3K27me3 antibody at 0, 12, 24 and 36 h after rescue of WT EED expression in i-WT-r mESCs. Staining at 36 h is shown at lower exposures. The rightmost panel is a zoomed-in image of the cell labeled with a red arrow. This figure has been modified from Oksuz et al., 201813. Please click here to view a larger version of this figure.
