The protocol for establishing primary mouse MD astrocytes outlined above should yield reproducible, high quality cultures. Although cultures initially contain a mix of astrocytes, fibroblasts, and other glial cells, including microglia and oligodendrocytes (Figure 1Bi,Biv; red arrowheads), the addition of AraC to the mixed culture between DIV5-DIV7 minimizes the proliferation of these contaminant cells. The combined AraC treatment and shaking-based purification strategy enriches the purity of the astrocyte cultures (Figure 1Bii) over traditional protocols that only include the purification steps (Figure 1Bv; blue arrowheads)13. The expected morphology and composition (assessed by GFAP and 4',6-diamidino-2-phenylindole [DAPI] staining) of the purified astrocyte cultures treated with AraC or left untreated is shown in Figure 1Biii,Bvi. Quantification of the percentage of the GFAP+ cells per field of view (ratio of number of cells with GFAP staining to total number of cells identified by DAPI staining) shows an increase of over 27% in purity with AraC supplementation (Figure 1C). This high-purity mouse astrocyte culture is suitable for evaluation of RNA and protein expression, cell morphology, and other functional assays.
We used lipofection-based transfection to express GFP-tagged versions of the gap junction protein Cx43 and the EAAT1 transporter in murine astrocytes. This methodology allows transient expression of proteins at levels that are optimal for live cell imaging without causing toxicity or affecting astrocyte viability (Figure 2A,E). Similarly, the use of a fluorescent probe permitted rapid and efficient labelling of acidic endo-lysosomal organelles (Figure 2I) to track their dynamics in astrocytes.
Figure 2 shows representative results of the analyses of cargo dynamics in astrocytes transiently transfected with EAAT1-GFP, Cx43-GFP, or labeled with a selective dye that recognizes acidic endolysosomal vesicles. Each of these cargos appeared as a fluorescent punctum decorating the perinuclear region, cytosol, and processes of astrocytes (Figure 2A,E,I, left panels). The time-lapse data was used to generate kymographs that track cargo motion in time and space (Figure 2A,E,I, right panels). In these kymographs, anterograde and retrograde movement of the indicated cargo is represented by trajectories with negative (green lines) and positive (red lines) slopes, respectively. Stationary vesicles are shown as vertical trajectories (blue lines). Kymograph analysis revealed that the three types of cargo analyzed undergo bidirectional transport with occasional fast, processive runs in both directions. Furthermore, quantification of the flux of a cargo through an area of the astrocyte (red box) revealed differences in the percentage of motile particles among cargos. For example, while 70% of EAAT1-GFP puncta are stationary (Figure 2B), less than 20% of Cx43-GFP (Figure 2F) and 45% of probe-labeled endolysosomal cargos (Figure 2J) are non-motile. These differences in motility among cargos are likely representative of their normal, baseline motility in the region of the astrocyte where the movies were acquired.
The precise mapping of the change in X-Y position along the full time scale for each particle obtained from the kymograph can also be used to evaluate other motion parameters, such as cargo velocity (Figure 2C,G,K) and run length (distance traveled) (Figure 2D,H,L). Motion parameters can be further analyzed for individual cargos to determine changes in directionality of movement (anterograde vs retrograde motion) as well as reversals in particle direction of motion, number of pauses, etc. The results from these types of analyses can provide meaningful quantitative information regarding changes in the cellular distribution of organelles and membrane proteins in astrocytes under basal or abnormal conditions in the intra- and extracellular environment.
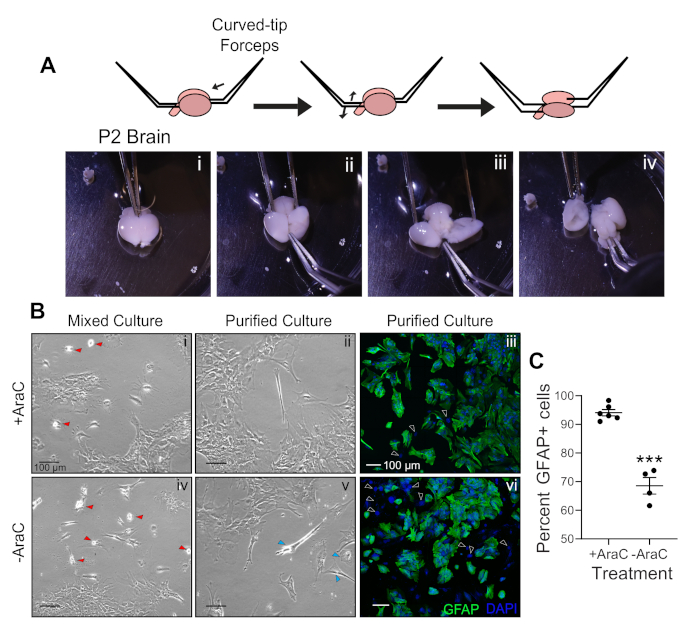
Figure 1: Establishment of primary mouse astrocyte cultures.
(A) Steps required for dissection of P2−P4 mouse brains. (B) (i,iv) Brightfield images of mixed glial cultures treated (i) and non-treated (iv) with AraC. Red arrowheads indicate contaminant microglia. (ii,v) Images of purified astrocyte cultures treated (ii) and non-treated (v) with AraC. Blue arrowheads indicate contaminant oligodendrocytes. (iii,vi) Confocal images of purified astrocyte cultures treated (iii) and non-treated (vi) with AraC. Green shows GFAP staining and DAPI (blue) labels all nuclei. (C) Percentage of GFAP-positive cells. Data represents mean ± SEM. ***p < 0.001, unpaired t-test. Please click here to view a larger version of this figure.
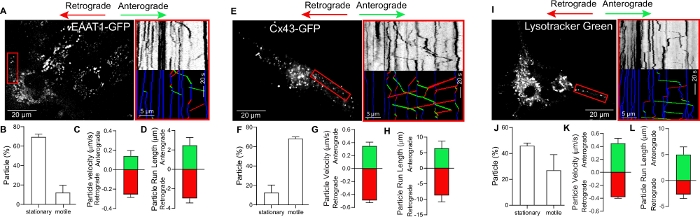
Figure 2: Quantitative analyses of cargo dynamics in astrocytes.
(A,E,I) Left panels: Astrocytes expressing EAAT1-GFP (A), Cx43-GFP (E) or treated with a probe that labels late endosomes and lysosomes (I). Red boxes indicate regions used to analyze particle dynamics. Right panels: Original and color-coded kymographs showing trajectories for the indicated cargos. Red lines represent retrograde-moving cargos, green lines anterograde-moving cargos, and blue lines non-motile cargos. (B,F,J) Percentage of stationary and motile particles. (C,G,K) Quantification of anterograde and retrograde velocity and (D,H,L) run length. Data represents mean ± SEM. Please click here to view a larger version of this figure.
