1. Generation of stable cell lines expressing 6His-Flag-Ubl
NOTE: Co-transfection of HEK-293T cells with pCCL-6HF-Ubl, pVSVG and delta-Helper.
- Day 0: Seed 293T cells in a 6-well plate to obtain 50-70% confluence the day after.
- Day 1: Co-transfect 50-70% confluent cells with a mix of 1 µg of pCCL-6HF-Ubl or pCCL-GFP, 1 µg of pVSVG and 1 µg of delta-Helper vectors, using a transfection reagent and protocol for lentivirus production. After 6 h of transfection, change the medium to a fresh one corresponding to cells to be transduced. Seed the cells to be transduced in a 6 well plate in order to obtain a 10-20% confluence the day after (the day of starting the transduction).
- Day 2: 24 h after transfection, recover the medium containing lentiviral particles and filter using 0.45 µm filters. If needed, add fresh medium at this point in order to produce a second batch of lentiviruses. Replace the medium of cells to be transduced (10-20% confluence) by the one containing lentiviruses.
NOTE: Lentiviral medium can be kept at +4 °C for several days before transduction or stored at -80 °C for months. - Incubate the cells with lentiviruses between 24 h to 72 h in a standard incubator (37 °C, 5% CO2), and then change the medium for fresh standard one. If possible, check GFP expression using an inverted fluorescent microscope to evaluate efficiency of transduction: percentage of expressing cells and relative level of expression per cell. If no fluorescence is detected, wait for additional 2-3 days as expression may take longer depending on cells type to be transduced.
- If GFP control is positive, grow all cells until having enough to perform an expression control of 6HF-Ubl by immunofluorescence and Western blot using anti-Flag antibody.
2. Double purification of modified proteins
NOTE: Buffer 1: 6 M Guanidinium-HCl, 0.1 M Na2HPO4/NaH2PO4, pH 8.0, 0.5% Triton X-100.
Buffer 2: 50 mM NaH2PO4, 150 mM NaCl, 1% Tween20, 5% Glycerol, pH 8.0.
Buffer 3: 100 mM NH4HCO3, pH 8.0.
- Cell lysis: Once ready, wash culture dishes at least one time with phosphate-buffered saline (PBS) at room temperature (RT) and proceed to cell lysis or, alternatively, flash freeze in liquid N2 and store at -80 °C. For lysis, add 2 mL of Buffer 1 per a 15 cm dish at RT. Use a cell scraper to recover all lysates in 50 mL conical centrifuge tubes (final volume about 20 mL).
- Sonicate the lysates three times for 30 s separated by a 1 min pause.
- Centrifuge the sonicated lysates at 15,000 x g for 15 min.
- Transfer the supernatant to a new tube using a cell strainer (40 µm).
- Determine samples' concentration and adjust, if necessary, to obtain the same amount of proteins and same volume. Use a total amount of protein between 50 and 100 mg (10 dishes with 15 cm diameter for MiaPaCa-2 cells).
- Add Ni2+-NTA beads, using 2 µL of beads per 1 mg of protein.
- Rotate at 30 rpm during 2.5 h at RT.
- Pellet the beads at 500 x g for 5 min.
- Wash the beads with 1 mL of Buffer 1, transferring the samples to a 1.5 mL microcentrifuge tube, then transfer the tubes on ice. Perform all the next steps on ice or at 4 °C.
- Wash two times with 1 mL of ice-cold Buffer 2 containing 10 mM imidazole.
- To elute bound proteins, add 600 µL of Buffer 2 containing 250 mM imidazole and rotate for 2 h at 4 °C.
- Pellet the beads by centrifugation at 500 x g for 1 min. Transfer the supernatants to new, pre-cooled, 1.5 mL tubes and add 50 µL of anti-Flag M2 antibody conjugated beads.
- Rotate at 30 rpm for 2.5 h at 4 °C, then wash 2 times with 500 µL of Buffer 2, then 2 times with 500 µL of Buffer 3.
- For the final elution, add 100 µL of Buffer 3 containing a Flag peptide at 0.1 µg/µL and rotate at 4 °C for 1.5 h.
- Centrifuge at 500 x g for 1 min and transfer the supernatants to new pre-cooled tubes.
- Take 10% (10 µL) to load on SDS-PAGE and perform a silver staining of the gel to control the purification quality. If the purification looks good, analyze the 90% left by LC-MS/MS.
3. Processing of mass spectrometry data to generate profiles of Ub/Ubls PTMs and to identify significant differences between them
NOTE: Results from MS analysis contain many information including the total number of peptides as well as the peak area values (mean of TOP 3 peptide area10) for each protein identified in each samples. These data can be processed using either the peptide count numbers or the peak area values, or both. For calculation with peak areas, because these values are usually in the range of 106, it is necessary to divide them by this order before applying the same formulas as below. The results obtained with both methodologies of counting should show a strong correlation as it usually does. For each identified protein, use the following formulas where:
v1  peptides values in non-treated ubiquitin sample (e.g., Ub – drug)
peptides values in non-treated ubiquitin sample (e.g., Ub – drug)
v2 peptides values in Gemcitabine treated ubiquitin sample (e.g., Ub + drug)
k1 peptides values in non-treated control GFP sample (e.g., GFP – drug)
k2 peptides values in Gemcitabine treated control GFP sample (e.g., GFP + drug).
- Normalization: Normalize values between drug treated cells and untreated cells for Ubiquitin and GFP using the following formulas. Normalized v = V and normalized k = K.
V1=v1.(∑v1+ ∑v2) / (2. ∑v1) ; V2=v2.(∑v1+ ∑v2) / (2. ∑v2)
K1=k1.(∑k1+ ∑k2) / (2. ∑k1) ; K2=k2.(∑k1+ ∑k2) / (2. ∑k2) - Removal of background: Using the following formulas, subtract values in control sample (GFP) from values in the ubiquitin sample to obtain specific values (V'1 and V'2) for each identified protein in both conditions.
V'1=V1-K1 if V1-K1≥0 ; V'1=0 if V1-K1<0
V'2=V2-K2 if V2-K2≥0 ; V'2 =0 if V2-K2<0 - Variation (Var) of ubiquitination. To obtain a score (between -100 and +100) for positive and negative variations of PTMs induced by a drug, use the following formula in which the difference between specific values of treated and untreated samples are divided by the sum of all values, including those in control (to penalize proteins also identified in control GFP), and multiply by 100.
Var = (V'2-V'1)/(V1+K1+V2+K2)*100 ; -100<Var<100 ;
Variations below -50 (repression of PTM) or above 50 (induction of PTM) are usually considered as significant. - Confidence (Conf). Use the following formula to obtain a confidence value between 0 and 100%,:
Conf = ((V1+V2)2/(1+V1+V2+K1+K2)2)*100 – 100/(1+V'1+V'2) ; =0 if <0
Values above 50 are usually considered to be confident. - To obtain a nicer distribution of induction/repression values and to consider both variation and confidence parameters, multiply Var and Conf values using the following formula where V Var and C Conf
=SI(V2>0;((V2*C2)^2)/(10^6);-((V2*C2)^2)/(10^6))
NOTE: As peak area values are usually more accurate than peptide counting, it is possible to use specific software which are dedicated to the interpretation of this kind of data such as Perseus (https://www.biochem.mpg.de/5111810/perseus), following recommendations of use.
Transduction of culture mammalian cells to create GFP and 6HF-Ub expressing cells
To produce lentiviruses which will be used later to transduce MiaPaCa-2 cells, 70% confluent HEK-293T cells are co-transfected with an equal amount of the three vectors, pCCL-6HF-Ubiquitin or GFP/Delta-Helper/pvSvG. After 24 h of production, the medium containing lentiviral particles is recovered and filtered. It is possible at this point to control the efficiency of the transfection by checking the green fluorescence of GFP expressing 293T cells on an inverted microscope. It should be near 100% of cells. MiaPaCa-2 cells are incubated with lentiviral supernatant for 1 to 3 days. The efficacy of lentiviral transduction is first controlled by looking at the GFP fluorescence of GFP transduced cells (Figure 1A upper panel). The GFP expression level may vary from one cell to another but 100% of them should be fluorescent. Once this control is done, the expression of Flag-ubiquitin will have to be also controlled. This is done by immunofluorescence staining using an anti-Flag antibody (usually M2 monoclonal) (Figure 1A lower panel). This will show the percentage of transduced cells, which should be 100% to guarantee a stable expression over future passages of cell culture. In order to control the expression level of exogenous Flag-ubiquitin, lysates from transduced cells are analyzed by SDS-PAGE followed by Western blot with anti-Flag antibody (Figure 1B). Both cell lines can be frozen.
Two steps purification and control by SDS PAGE and silver staining
Once the stable expressing cell lines have been validated, both GFP and 6HF-ubiquitin cells are amplified until enough material is obtained in order to proceed with the two-step purification. It is recommended to keep a backup of these cell lines as frozen stock in liquid nitrogen in order to be able to thaw them when needed. 36 h before processing, half of the cells (half GFP and half 6HF-Ubiquitin) are treated with 10 µM of Gemcitabine. When ready, ubiquitinated proteins are purified from 6HF-ubiquitin and from GFP control cells, using the two-step purification protocol (Figure 2A). 10% of the final elution is used to control the amount and integrity of purified material by SDS-PAGE and silver staining of the gel (Figure 2B). Molecular weight markers bands can be used to estimate the amount of purified ubiquitinated proteins. Alternatively, a known amount of BSA or any other protein can be loaded in a line of the gel to help quantifying the purified proteins. Once this verification is done, the remaining 90% of samples are analyzed by liquid chromatography coupled to tandem mass spectrometry to allow identification and semi-quantitation of purified proteins.
Identification of ubiquitinated proteins by background (GFP samples) subtraction
Data from LC-MS/MS analysis of the samples give the names and quantifications (peak areas and number of observed peptides) of each identified protein in GFP and 6HF-Ub samples. The proteins having the highest quantification in ubiquitin sample compared to GFP sample are most likely to be the ones really ubiquitinated and applying the formulas described in methods above allows their classification by giving a confidence score from 0 to 100%. Proteins identified with a score above 50% are considered ubiquitinated ones (Figure 3A).
This step which basically removes background proteins (GFP) from ubiquitin samples led to the identification of 364 proteins significantly ubiquitinated (Figure 3B)7. Note here that the proteins identified with the highest scores are mostly already known as main targets of ubiquitination which proves the efficacy of this purification method.
Then it is possible to perform a gene set enrichment analysis (GSEA) of these ubiquitinated proteins in order to highlight the biological processes in which they are involved (Figure 3C), their molecular functions, their cellular compartment, or any other gene ontology categorization. It is interesting to compare this ubiquitinated proteome with the full proteome of the cell when possible. Indeed, this analysis reveals the real contribution of these ubiquitinated proteins in specific processes such as translation or proteolysis for example (Figure 3C).
The main purpose of this kind of experiment is to identify alterations within the ubiquitinated proteome induced by a treatment for example, here gemcitabine. By comparing the ubiquitinome (the ubiquitinated specific proteome) in treated and untreated cells using the specific formula yields a value from -100 (repression of ubiquitination) to +100 (induction of ubiquitination). Considering only the values below -50 or above +50 as significant, a total of 73 induced ubiquitinations and 29 repressed ubiquitinations have been identified (Figure 4A). GSEA analysis of these alterations of ubiquitination revealed specific enrichments in DNA repair processes or cell cycle, and important variations in translation and RNA metabolic processes (Figure 4B). This result is highly logical since gemcitabine is a base analogue that blocks DNA synthesis and provokes DNA damages.
To go further, it is also interesting to use databases of interacting proteins in order to explore and validate potential interacting networks formed by gemcitabine induced alterations of ubiquitination (Figure 5). This led to the identification of functional interacting networks strongly affected by increased or decreased ubiquitination of the involved proteins.
Finally, among the altered ubiquitination, some may involve proteins of highest interest, due to their known or potential functions according to literature. Hence, one important step is to validate that what is observed by mass spectrometry is real and trustworthy. PCNA was one of the most over-ubiquitinated protein upon gemcitabine treatment detected by mass spectrometry analysis (Figure 4A). To verify that gemcitabine indeed induces the ubiquitination of PCNA, MiaPaCa-2 cells expressing 6HF-Ub and GFP are grown, treated or not, and subject to either Ni-NTA purification in denaturing conditions or to anti-Flag immunoprecipitation followed by Flag peptide elution in native conditions, resolved on SDS PAGE followed by western blot with the corresponding antibody. The result shown in Figure 6 confirmed that indeed PCNA is strongly ubiquitinated in response to gemcitabine treatment in MiaPaCa-2 cells.
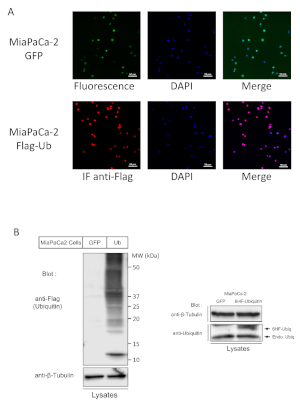
Figure 1: Establishment of stable cell lines expressing 6His-Flag-Ubl. (A) Control of GFP expression in GFP transduced cells (upper panel) and of 6His-Flag-Ubiquitin by immunofluorescence using anti-Flag (M2) antibody as primary and alexa-567 anti-mouse secondary antibody. DAPI was used to stain nuclei. Scale bar: 50 µm. (B) Control of 6His-Flag-Ubiquitin expression in cell lysates by Western blot using anti-Flag antibody (Alternatively, an anti-6his antibody can be used) on the left, and anti-ubiquitin antibody, on the right, to compare the expression of 6His-Flag-Ubiquitin (6HF-Ubiq) with endogenous Ubiquitin (Endo. Ubiq). Please click here to view a larger version of this figure.
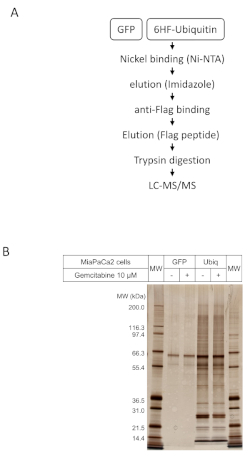
Figure 2: Two step purification of ubiquitinated proteins. (A) Schematic representation of the procedure. (B) 10% of the final elution was subjected to SDS PAGE followed by silver staining of the gel in order to estimate the quantity and purity (compared to GFP) of isolated proteins. Please click here to view a larger version of this figure.
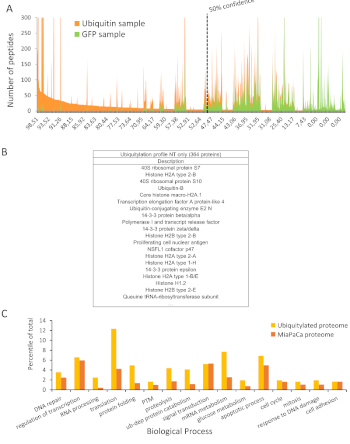
Figure 3: Identification of ubiquitinated proteins (adapted from Bonacci et al.7). (A) Relative amount of specific (ubiquitin sample) and non-specific (GFP sample) peptides for each identified protein were plotted in function of their confident scores. As shown, the proportion of non-specific over ubiquitinated proteins becomes too important below the score of 50. Hence, only proteins identified with a score superior to 50 are considered significant. (B) Table showing the 20 best ubiquitinated proteins among the 364-total identified. (C) Repartition of ubiquitinated proteins and total proteins of MiaPaCa-2 cells within biological processes (Values > 1.5% were considered only). Please click here to view a larger version of this figure.
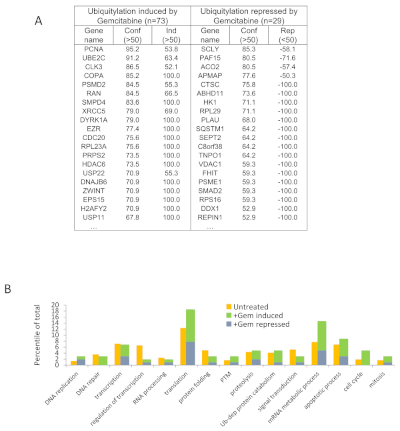
Figure 4: Gemcitabine induced alterations of PTM profiles (adapted from Bonacci et al.7). (A) Listing of the 20 proteins with highest increased (total 73) or decreased (total 29) ubiquitination upon gemcitabine treatment (Conf: confidence; Ind: induction; Rep: repression). (B) Repartition of Gemcitabine induced altered ubiquitination within biological processes and comparison with non-treated. Please click here to view a larger version of this figure.
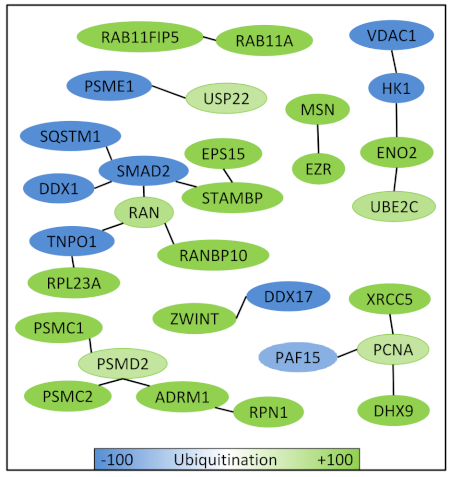
Figure 5: Functional interactomes of gemcitabine induced altered ubiquitination. Potential interactions between all proteins with gemcitabine induced alteration of ubiquitination are identified using a protein-protein interactions database (STRING: string-db.org). Please click here to view a larger version of this figure.
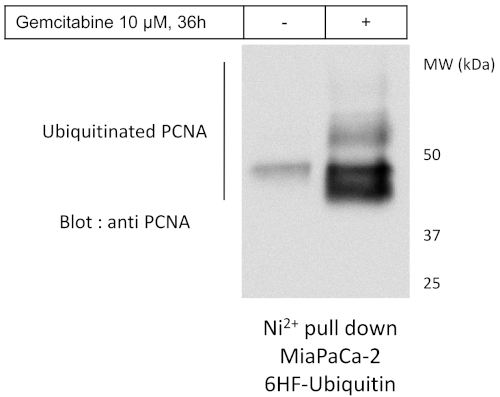
Figure 6: Biochemical validations of interesting gemcitabine induced alterations of ubiquitination. In order to validate the increased ubiquitination of PCNA after gemcitabine treatment, lysates of cells expressing 6HF-Ubiquitin, treated or not with gemcitabine, were subjected to Nickel pull-down (Ni-NTA) followed by anti-PCNA Western blot. Please click here to view a larger version of this figure.
