Informed patient consent and full compliance with relevant national ethics requirements in each jurisdiction e.g., the Human Tissues Act (HTA – United Kingdom, 2004) and the Health Insurance Portability and Accountability Act (HIPAA – United States, 1996) are mandatory. Be certain to have fully documented ethics approval before beginning any work on human-derived materials. Blood used in the optimization of this protocol was provided with appropriate consent from the Volunteers Advancing Medicine Panel (VAMP) (ethics reference 16/EE/0459, study CRF494, sub study 001) run by the NIHR Cambridge Clinical Research Facility, Cambridge, United Kingdom, under a Human Biological Samples supply agreement with the National Institute for Health Research (NIHR) Cambridge Biomedical Research Centre. The AstraZeneca Biobank in the UK is licensed by the Human Tissue Authority (License No. 12109) and has National Research Ethics Service Committee (NREC) Approval as a Research Tissue Bank (RTB) (REC No 17/NW/0207).
1. General preparation guidance
NOTE: All work with unfixed human material such as the blood, plasma and PBMCs in this protocol must operate under the assumption that these materials may carry potentially infectious agents and so must be performed under suitable biosafety precautions. For patients that have been tested as negative for known pathogens, do not assume samples are non-infectious and so suitable safety precautions must still be applied. Waste products generated from these materials should be treated with the same biosafety precautions and disposed according to local rules.
- Choose between the two types of mononuclear cell preparation tubes that utilize either sodium citrate or sodium heparin as anticoagulants. For many downstream applications these two anticoagulants can be used interchangeably but both anti-coagulants should be tested before selecting one for the trial.
- Label one 8 mL mononuclear cell preparation tube to be used for the blood collection with the patient’s coded ID. Keep the tubes at room temperature (18-25 °C).
- Store 1x PBS at room temperature (18-25 °C). Use 30 mL per preparation.
- Make sure tubes for separate plasma samples and the cryovial for the PBMC sample are accurately labeled with unique identifiers, as specified in the appropriate section of the lab manual.
- If PBMCs are going to be used to monitor phosphorylated proteins, prepare PBS supplemented with phosphatase inhibitors: mix 5 mL of PBS with 50 µL of phosphatase inhibitor cocktail 2 + 50 µL of phosphatase inhibitor cocktail 3. Prepare fresh and keep on ice until use.
- If PBMCs are going to be cryopreserved, prepare 1 mL freezing mixture per sample by mixing 90% FBS + 10% DMSO.
2. PBMC collection (Figure 1A)
- Draw 8 mL blood into the mononuclear cell preparation tube using the standard technique described by the manufacturer. Invert the tube gently 8 to 10 times to mix the anticoagulant additive with blood. Do not shake to avoid hemolysis. Record the time at which the blood was drawn.
- After collection, store the tube upright at room temperature until centrifugation. Process the samples as soon as possible, ideally within one hour of this blood collection but not later than 4 h post collection. Record the timing when starting the processing of the blood.
- Immediately prior to the centrifugation remix the blood sample by gently inverting the tube 8 to 10 more times.
- Centrifuge the tube/blood sample tubes in a horizontal rotor (swing-out head) at 1,500 – 1,800 x g for 30 min at room temperature (18-25 °C). Ensure that all the tubes are balanced properly.
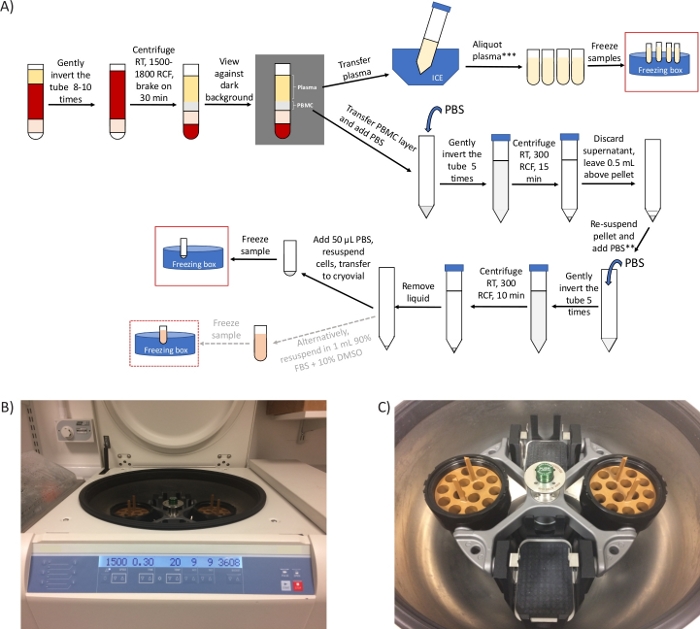
Figure 1: General overview of the protocol and representative images of the centrifuge. (A) Schematic overview of the protocol for the preparation of PBMCs and plasma. *If there are time constraints, step 2.4 can be shortened to 20 min and steps 3.3 to 3.6 can be removed. ** Take an aliquot to count cells, if required. *** 2 extra centrifugation steps required for ctDNA analysis/metabolomics (B) image of a swing-out head rotor centrifuge set for step 2.4. (C) Image of the rotor, which includes two buckets containing the adaptors to spin mononuclear cell preparation tubes. Please click here to view a larger version of this figure.
NOTE: x g (also referred to as RCF, relative centrifugal units) and RPM (revolutions per minute) are different units. Be sure to set the centrifuge for x g (RCF). Use the online calculator (see Table of Materials) for the conversion. If only fixed-angle rotor is available, perform this step for 10 min at 1,500 – 1,800 x g.
- After centrifugation, check for the presence of a dark red layer under the barrier (containing mostly red blood cells), and 2 layers above the barrier. The top layer is the plasma (straw-colored) and the whitish layer underneath is the buffy coat containing the PBMCs. These layers can be easily distinguished when viewing the tube against a dark/black background.
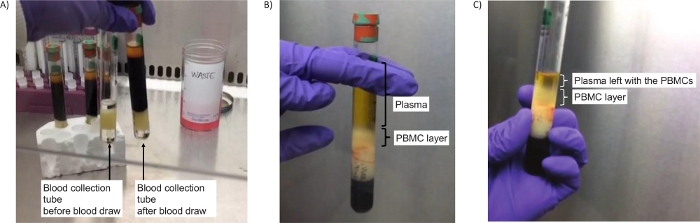
Figure 2: Tubes to isolate PBMCs. (A) Image of the tubes before centrifugation (step 2.4), either empty (left) or containing blood (right). (B) Successful separation of the PBMC layer after centrifugation (step 2.5). (C) Image of the PBMC layer after centrifugation and a bit of plasma left to ensure all PBMCs are collected (step 2.7). Please click here to view a larger version of this figure.
NOTE: If these layers are not visible, this probably indicates an error in setting up the centrifugation units. Ensure the right centrifuge adapter is used and the correct “x g” are set. Repeat step 2.4.
- Immediately following centrifugation, use a serological pipette to transfer approximately half of the plasma into a labeled 15 mL size conical centrifuge tube with cap, while being careful to not disturb the PBMC layer (approximately 4-5 mL). Temporarily store this tube with plasma on wet ice to be used later in the step 4. Set aside for now.
- Collect the entire PBMC layer with a Pasteur pipette by placing the pipette within the layer of cells and transfer to a different 15 mL size conical centrifuge tube with cap. It is acceptable to also take a small amount of plasma, if necessary, to completely get all the PBMC layer. The volume is usually 1-2 mL. Immediately continue with the step 3 below.
3. PBMC washing steps
NOTE: The purpose of the wash steps is to dilute out and remove residual platelets and plasma from the PBMC pellet. All centrifugation steps should be performed at room temperature (18-25 °C).
- Add room temperature PBS to the PBMC tube to bring the volume to 15 mL. Cap the tube. Mix cells by gently inverting the tube 5 times.
- Centrifuge for 15 min at 300 x g. Note that this is a much gentler centrifugation than the initial centrifugation.
- Visualize the pellet by viewing the tube against a dark/black background. Remove the supernatant by vacuum aspiration or with a pipette (Figure 3A). Discard the supernatant leaving a volume of approximately 500 μL above the whitish colored PBMC pellet.
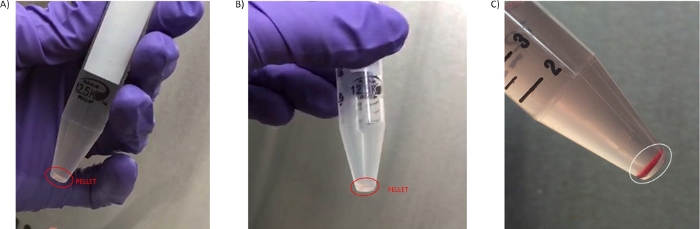
Figure 3: PBMC pellets. (A) Pellet obtained in the first wash step 3.3. (B) Pellet isolated in the second wash (step 3.7). (C) Pellet with high level of hemolysis obtained from blood processed later than 4 h from the blood draw isolated in the second wash (step 3.6). Please click here to view a larger version of this figure.
- Resuspend the cell pellet by gently pipetting up and down in the residual supernatant.
- Add additional 1x PBS to bring the volume to 10 mL. Cap the tube. Mix the cells by inverting the tube gently 5x. If cell counting is required in the study protocol go to step 3.5.1, if not required move directly to step 3.6.
- Take a 40 μL aliquot of the re-suspended cells and mix with 40 μL of trypan blue. Count the viable cells using a hemocytometer or an automatic cell counter.
- Centrifuge the suspension from step 3.5 for 10 min at 300 x g. Remove as much supernatant as is reasonably possible without disturbing the cell pellet.
- Carefully remove and discard any remaining supernatant above the cell pellet by pipetting using a fine tip Pasteur pipette or a micropipette without disturbing the cell pellet. Leave little to no liquid above the pellet after completing this step. If the endpoint of this protocol is a PBMC pellet move to step 3.8; if the endpoint is cryopreserved PBMCs go to step 3.10.
- Add 50 µL of new PBS (or the supplemented PBS prepared in step 1.5 if applicable to your endpoint) to the pellet and pipette up and down gently using a micropipette to homogenously resuspend all the cells. Transfer the entire cell suspension to a labeled 1.5 mL cryovial and immediately place on wet ice until freezing the samples.
- Freeze the labeled samples by placing them in liquid nitrogen or directly in dry ice. Cells can also be frozen at -80 °C freezer using cell freezing boxes. In this case, prechill the boxes completely in -80 °C before adding the cell tubes. Record the time at which cell pellets were frozen. Once cells freeze, store at -80 °C, ship frozen on dry ice.
- Optionally, to cryopreserve the PBMCs re-suspend the pellet in 1 mL of the freezing mixture (step 1.6) and transfer to a labeled cryovial. Continue as described in step 3.9, but, in this case, it is only acceptable to use cell freezing boxes to freeze down the samples. Transfer to liquid nitrogen for shipping and storage after being in the freezing box for a minimum of 24 h.
4. Plasma preparation steps (Figure 1A)
- Following -80 °C storage of the PBMC pellets, now prepare the plasma aliquot that was temporarily stored on wet ice in step 2.6. Perform the following centrifugation steps to clarify the plasma if the purpose of the sample is ctDNA analysis, otherwise move directly to step 4.2.
- Centrifuge the plasma for 10 min at 1,600 – 2,000 x g at 4 – 8 °C in a fixed-angle rotor (or 15 min in a swing-out rotor).
- Carefully pipette off the plasma supernatant, taking care not to disturb any pellet and transfer to a new 15 mL tube. Discard the tube containing the pelleted material.
- Centrifuge the plasma for 10 min at 1,600 – 2,000 x g at 4 – 8 °C in a fixed-angle rotor (or 15 min in a swing-out rotor).
- Carefully transfer the plasma supernatant to a new 15 mL tube, being sure not to disturb any pelleted material. Discard the tube containing the pelleted material.
NOTE: If a refrigerated centrifuge is not available, keep cells on ice for 5 min between each spin, or centrifuge samples in a cold room.
- Transfer plasma in 1 mL aliquots into 5 fresh 2 mL microtubes. Use fewer than 5 vials if there is not enough plasma to fill 5 vials with 1 mL aliquots and it is expected that the last vial will contain less than 1 mL plasma. If there is more than 5 mL plasma, discard the remainder. Record the total volume of plasma in each tube.
- Immediately freeze the plasma aliquots upright, by storing them at -80 °C.
5. Sample shipment
- Ship both PBMC pellets and plasma samples on dry ice.
- Ensure that the sample is not thawed before and during shipment. Pack sufficient dry ice with the samples to ensure they remain frozen for the entirety of the shipping process, considering possible delays that may occur in transit.
6. Whole blood irradiation and western blot analysis of DDR biomarkers (Figure 4A)
NOTE: This is an ex vivo treatment that will not be required in most cases in the clinic, but these experiments are a valuable exploratory strategy to find suitable clinical biomarkers. Blood samples should be treated as soon as possible upon collection to ensure best results.
- Warm up the X-ray cabinet. Label three 15 mL tubes as 0, 0.2 and 7 Gy, respectively.
- Take three mononuclear cell preparation tubes containing freshly drawn blood from a single individual and after gently inverting the tubes 8 to 10 times transfer the bloods to the three 15 mL tubes.
- Place the 0.2 Gy tube in the X-ray cabinet, close the door and apply 0.2 Gy dose to the tube by selecting the shelf number and dose. Apply 7 Gy radiation dose to the 7 Gy tube by adjusting the dose selected in the cabinet.
- Incubate the three tubes at 37 °C for one hour.
- Transfer the blood back to the mononuclear cell preparation tubes and carry out the PBMC preparation protocol as for a clinical setting from step 2.4 to step 3.8.
- Add a volume of lysis buffer supplemented with protease and phosphatase inhibitors equal to the cell pellet volume (in this case 70 μL of RIPA buffer) to each of the cell pellets, pipette up and down and incubate on ice for 10 min. Sonicate the samples if a sonicator is available (3 cycles, 30 s ON / 30 s OFF, 4 °C), alternatively syringe the samples to break the nucleic acids to eliminate viscosity in the sample.
- Centrifuge the samples for 10 min at ≥ 15,000 x g, at 4 °C. Transfer each supernatant to a new 1.5 mL tube, qualitatively assess the level of hemolysis (visual inspection) and measure the protein concentration by any preferred method.
- Mix 40 μg of total lysate with sample loading buffer containing SDS and sample reducing agent. Boil samples for 5 min in a heat block.
- Load 20 μg of each sample per lane in duplicate in a 4-12% bis-tris protein gel and run an SDS-PAGE.
- After separation, transfer the proteins to a nitrocellulose membrane using a commercially available system (20 V, 10 min) and block with 5 % milk in TBST.
- Cut the membrane at the relevant molecular sizes and incubate with the primary antibodies overnight at 4 °C (see Table of Materials for antibodies and dilutions).
- Remove the primary antibodies and wash the membranes 3 times with TBST for 5 minutes at room temperature. Incubate with the HRP-conjugated secondary antibodies for 45 minutes at room temperature.
- Wash 3 times with TBST for 5 min.
- Apply the ECL reagent and analyze the images obtained relative to HRP signal.
To improve the quality of PBMC preparations in our clinical trials, we have generated a protocol with concise, clear steps that can be followed by hospital laboratory professionals, independent of their molecular biology background and laboratory skills. We have adapted the manufacturer’s protocol incorporating modifications on those steps where execution issues have been identified or reported from clinical sites involved in various multi-center clinical trials. However, the protocol can be further optimized to meet specific requirements, such as time constraints in the clinical site or type of downstream analyses (see Supplementary File). We demonstrate that the DDR can be analyzed in PBMCs by looking at specific biomarkers upon DNA damage generated by radiation.
The most common queries we have received from clinical sites relate to the centrifugation steps, which directly impact being able to successfully isolate PBMCs and obtain the final PBMC pellet. Using the appropriate type of swing-out head rotor centrifuge (Figure 1B,C) is the key to the success of the protocol. However, when only a fixed-angle rotor centrifuge is available at the clinical site, we suggest carrying out step 2.4 in a fixed-angle rotor at the same RCF as for a swing-out head rotor but for only 10 minutes. This ensures that a PBMC layer is separated from the plasma, (see Figure 2B,C). While layering blood on a density gradient separation medium requires a brake-off centrifugation, blood in mononuclear cell preparation tubes can be centrifuged with brakes on due to the presence of a gel barrier in the tube that ensures the preservation of the PBMC layer separated from the denser blood components27,28.
At least 4 mL of high quality, non-diluted plasma were obtained from the centrifugation of blood in the 8 mL tube format, which could be further clarified for its use in specialized analyses such as ctDNA sequencing or metabolomics studies29,30 (optional steps 4.1.1-4.1.4). The amount of isolated PBMCs, size of the pellets and hemolysis or red blood cell contamination have been other concerns coming from clinical sites and experience analyzing samples. The number of cells obtained from 8 mL blood was variable depending on the patient and disease setting, but in general the pellet obtained is small in size, and of a transparent/white coloration (Figure 3A,B). Due to these characteristics, it is important to visualize the pellet against a dark background to avoid its accidental aspiration during the wash steps (2.5 and 3.3). Sometimes pellets can have some red coloration due to red blood cell contamination (Figure 3C), and this has a negative effect on the quality of the preparation. To avoid losing small pellets such as those shown in Figure 3, transferring the PBMC pellet to a cryovial is facilitated by step 3.7 where the PBMC pellet is resuspended in 50 µL of PBS.
The typical yield when using these tubes and blood from healthy individuals ranges between 7 to 21 x 106 cells for 8 mL, and a cell recovery between 70 and 80% as it is our experience and as it has been previously shown27,28. This depends on both the individual cell counts and the operator and it is comparable to the cell numbers and cell recovery values obtained by other methods using density gradient (including systems utilizing tubes with a separation barrier)15,27,28. An illustrative example of the variation on number of PBMCs isolated with this method depending on disease setting is the analysis of PBMC markers in chronic lymphocytic leukemia (CLL) patients. The number of cells recovered from an 8 mL mononuclear cell preparation tube when applying this protocol varied from 1.62 x 104 to 1.99 x 109 in 45 samples obtained from 7 patients in study NCT0332827331 (Table 1). A related parameter is the protein concentration of the PBMC lysates obtained by this method, and this depends on the number of cells isolated and the efficiency of the protein extraction. The cell pellets lysed in section 6 were generated with RIPA buffer and sonication (step 6.6). The resuspension of the cell pellets in their same volume of lysis buffer usually results in a range of concentrations from 3 to 10 mg/mL, and in this particular example the lysate concentrations were 6.8, 8.3 and 8.6 mg/mL for 0, 0.2 and 7 Gy, respectively. However, this is subjected to a high variability when receiving samples from clinical sites that have not been optimally prepared, patient’s disease, and the presence of hemoglobin from red blood cell contamination. For example, very small pellets need to be resuspended in a volume of lysis buffer larger than the cell pellet volume to allow for both protein concentration measurement and downstream biomarker analysis, resulting in more diluted samples. In such case, if a sample concentration is below 1 mg/mL this can pose a challenge to perform assays like western blot due to this high dilution factor. In contrast, in the CLL samples previously mentioned, the volume of lysis buffer added to resuspend the cell pellets varied from 50 to 500 μL, and protein concentration spanned from 1.62 to 19.77 mg/mL (Table 1).
When samples present red blood cell contamination or hemolysis (Figure 3C), the protein concentration of the PBMC lysate becomes overestimated due to the inclusion of hemoglobin from the erythrocytes. This is the reason why one should do a visual inspection and annotation of such samples, as described in step 6.7 of the protocol. Loading sample in excess can compensate for the presence of hemoglobin when performing biomarker analysis as far as a loading control is included in the assay. Other more quantitative methods to measure hemolysis could be implemented, such as measuring absorbance at 414 nm32.
The DDR was analyzed in PBMCs obtained following this clinical protocol. To illustrate a situation that mimics DNA damaging clinical treatments such as radiotherapy or chemotherapy, whole blood from healthy volunteers was ex vivo subjected to X-ray radiation (Figure 4A). Ionizing radiation (IR) such as X-rays induces different types of DNA damage, including DNA double-strand breaks (DSBs). DNA damage sensing of these lesions activates PIKKs such as ataxia-telangiectasia mutated (ATM), ATM and Rad3-related (ATR) and DNA-dependent protein kinase (DNA-PK), which engage DNA repair mechanisms like homologous recombination (HR) or non-homologous end joining (NHEJ). Activation of ATM by the presence of DSBs occurs by its recruitment to sites of damage by the MRN (MRE11-RAD50-NBS1) complex, causing ATM autophosphorylation at several residues including Ser1981. In turn, activated ATM phosphorylates the components of the MRN complex and other proteins such as histone variant H2AX on Ser139 (where pSer139-H2AX is also known as γH2AX) to promote a structural change in the chromatin spanning from the DSB which facilitates the recruitment of other DDR factors33. PBMCs are responsive to ionizing radiation despite their low proliferation rate and mass spectrometry methods have allowed quantification of the upregulation of phosphorylated Ser635 on RAD50 by ATM. This phosphorylation is reduced in the presence of ATM inhibitors and RAD50 pS635 has been further validated as a pharmacodynamic biomarker for clinical ATM inhibitor treatments in tumors by immunohistochemistry8. To evaluate the response of PBMCs to radiation, blood from healthy volunteers was subjected to different IR doses and samples were collected after a 1 h incubation at 37 °C (Figure 4A). For this purpose, bloods were transferred to plastic tubes to avoid reducing the yield of the PBMC isolation due to high temperature (step 6.2). We analyzed how ATM was activated not only by looking at the previously reported RAD50 pS635 but also ATM pS1981 and γH2AX. In the three cases examined an increase in these post-translational modifications was observed at higher IR doses (Figure 4B). Interestingly, the phosphorylation of ATM and RAD50 was substantial at the low dose of 0.2 Gy, which suggests these post-translational modifications may be feasibly interrogated as PD biomarkers for treatments involving the generation of DNA DSBs with a good dynamic range, not only in tumor samples but in peripheral blood. This allows the monitoring of the PD response to the treatment by acquiring longitudinal samples through the course of treatment. The timing from the blood draw to the processing of the samples is critical to ensure these signaling cascades are still active as delays in processing will impact on the kinetics of such cascades and one could miss the phosphorylation events used as phosphomarkers.
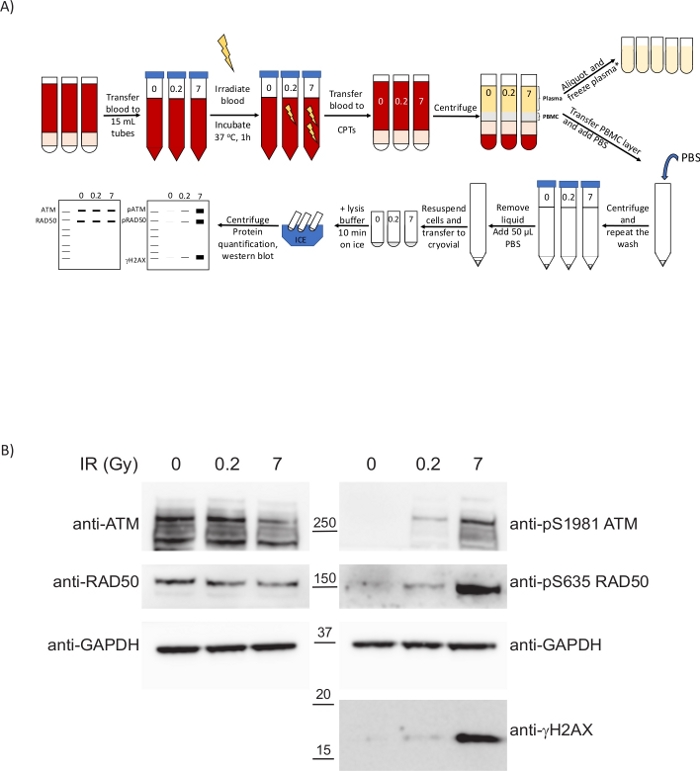
Figure 4: PBMCs isolated from ex vivo irradiated blood following the present clinical protocol display biomarkers to inform on the extent of the DNA damage caused by the treatment. (A) Schematic overview of the protocol for the preparation of PBMCs and analysis of the DDR upon whole-blood irradiation. *2 extra centrifugation steps required for ctDNA analysis/metabolomics. (B) Western blot showing dose-dependent upregulation of DDR phospho-biomarkers in PBMCs. Please click here to view a larger version of this figure.
| Sample | Number of PBMCs/ 8 mL blood | Supplemented RIPA buffer volume (μL) | Final concentration (mg/mL) |
| A.1 | 39100000 | 70 | 12.16 |
| A.2 | 97400000 | 100 | 4.54 |
| A.3 | 233000000 | 150 | 7.63 |
| A.4 | 316000000 | 150 | 16.87 |
| A.5 | 387000000 | 150 | 12.60 |
| A.6 | 459000000 | 150 | 12.71 |
| A.7 | 414000000 | 200 | 15.67 |
| A.8 | 253000000 | 150 | 14.56 |
| A.9 | 509000000 | 300 | 10.67 |
| B.1 | 15200000 | 70 | 2.96 |
| B.2 | 10500000 | 50 | 4.59 |
| B.3 | 13200000 | 50 | 3.99 |
| B.4 | 34800000 | 100 | 10.41 |
| B.5 | 1620000 | 70 | 7.11 |
| B.6 | 70200000 | 100 | 9.26 |
| B.7 | 65400000 | 100 | 12.10 |
| B.8 | 91100000 | 150 | 11.82 |
| C.1 | 6330000 | 70 | 4.04 |
| C.2 | 4400000 | 150 | 19.77 |
| C.3 | 68000000 | 100 | 8.96 |
| C.4 | 35100000 | 50 | 9.30 |
| C.5 | 35400000 | 70 | 10.55 |
| C.6 | 99200000 | 100 | 16.19 |
| D.1 | 402000000 | 70 | 7.23 |
| D.2 | 826000000 | 300 | 16.95 |
| D.3 | 1990000000 | 300 | 14.87 |
| D.4 | 1000000000 | 300 | 18.34 |
| D.5 | 1160000000 | 400 | 16.13 |
| D.6 | 806000000 | 400 | 19.40 |
| E.1 | 302000000 | 300 | 13.86 |
| E.2 | 990000000 | 500 | 19.04 |
| E.3 | 1200000000 | 400 | 17.13 |
| F.1 | 4010000 | 50 | 1.62 |
| F.2 | 5170000 | 50 | 2.84 |
| F.3 | 2810000 | 50 | 3.69 |
| F.4 | 3700000 | 75 | 3.62 |
| F.5 | 3460000 | 70 | 4.03 |
| F.6 | 7060000 | 50 | 3.32 |
| G.1 | 60700000 | 70 | 6.57 |
| G.2 | 82100000 | 150 | 7.78 |
| G.3 | 30500000 | 70 | 8.28 |
| G.4 | 134000000 | 100 | 15.14 |
| G.5 | 91900000 | 100 | 8.61 |
| G.6 | 372000000 | 150 | 15.88 |
| G.7 | 574000000 | 200 | 15.01 |
| Longitudinal PBMC samples corresponding to 7 patients | |||
Table 1: Cell number and protein concentration of PBMC pellets from chronic lymphocytic leukemia patients (CLL). The data presented in this table correspond to 45 PBMC samples from 7 CLL patients participating in study NCT03328273 collected in mononuclear cell preparation tubes. These are original data generated using samples from the cited study31.
Supplementary File: Alternative protocol options. Please click here to download this file.
