A complete set of experiment should include Cre-BFP only transfection as negative control, Cre-BFP plus 480 kDa ankyrin-G co-transfection as positive control and a non-transfected condition as technique control. In Cre-BFP only control, transfected neurons lack the accumulation of AIS markers, including ankyrin-G (ankG), beta4-spectrin (β4), neurofascin (Nf) and voltage gated sodium channels (VSVG) (Figure 4A)16. In contrast, Cre and 480 kDa ankyrin-G co-transfected neurons have fully assembled AIS revealed by the present of AIS markers (Figure 4B). It is important to confirm the quality of culture by comparing with the non-transfected dishes. Unhealthy neurons tend to show abnormal AIS structure, like discontinued or ectopic AIS (Figure 4C).
Then we showed an example of evaluating how an ankyrin-G human neurodevelopmental disorder mutation (ankG-K2864N) affects AIS assembly (Figure 5). 3 div ANK3-E22/23f/f neurons were transfected with Cre-BFP and wildtype 480 kDa ankyrin-G (ankG-WT) or 480 kDa ankyrin-G baring human mutation (ankG-K2864). Neurons were fixed at div7 and stained for ankyrin-G. Images were collected from 10-15 transfected neurons and 10-15 control neurons on the same coverslips and processed with maximum intensity projection. Then we draw a line at the AIS as shown and measure the mean intensity across the line. After averaging the AIS intensity, we plot the AIS intensity from the soma to the distal axon. AIS enriched protein normally showed a fast increase of signal from the proximal axon and a slow decrease of signal to the distal axon. AIS assembled by ankyrin-G with human mutant showed an increase and decrease of signal. But when aligned with the non-transfected AIS, the mutant curve is wider, and peak of the curve is lower suggesting a structure change of AIS. The wild type ankyrin-G assembled AIS closely aligned with the non-transfected one.
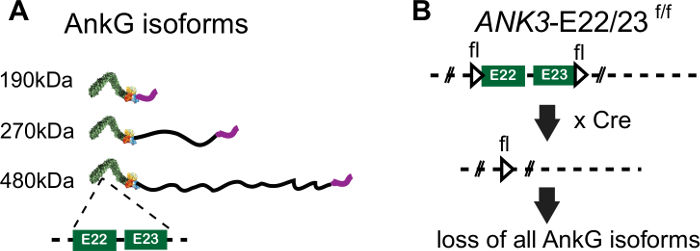
Figure 1: The genomic editing of ANK3-E22/23-flox. (A) Schematic representation of protein domains for 3 ankyrin-G isoforms. The location of exon 22 and 23 encoded regions in canonical domain is pointed by the dash line. (B) The position of LoxP sites in ANK3-E22/23-flox mice is indicated by triangle. In the present of Cre recombinase, exon 22 and 23 is deleted and causes loss the expression of all 3 isoforms of ankyrin-G. Please click here to view a larger version of this figure.
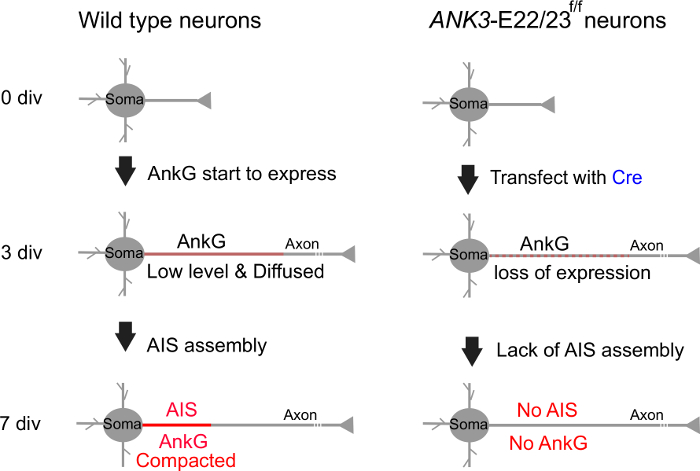
Figure 2: Loss of AIS in ANK3-E22/23-flox neurons in the present of Cre recombinase. A diagram shows the time frame of ankyrin-G expression and AIS assembly in wild type neurons versus in ANK3-E22/23f/f neurons with Cre transfection at 3 div. Please click here to view a larger version of this figure.

Figure 3: Workflow of protocol. Please click here to view a larger version of this figure.
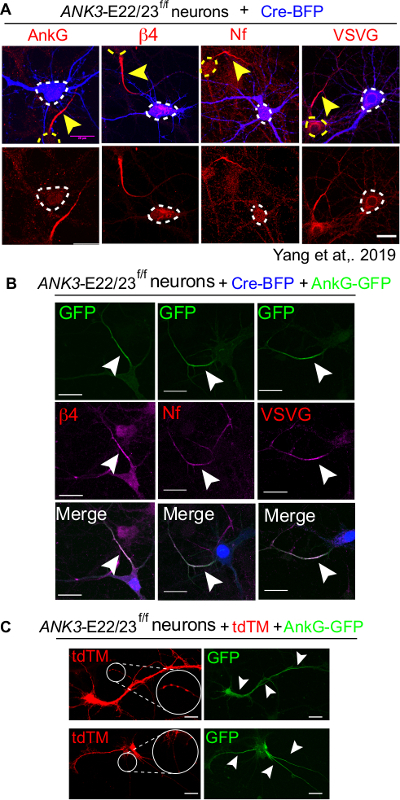
Figure 4: A full rescue of AIS by 480kDa AnkG in ANK3-E22/23-flox neurons transfected with Cre. 3 div neurons of ANK3-E22/23f/f mice were transfected with Cre-BFP (A) or with a Cre-BFP and wild type 480 kDa ankyrin-G-GFP (B). Neurons were fixed at 7 div and stained for ankyrin-G (ankG), β4-spectrin (β4), neurofascin (Nf) and voltage gated sodium channels (VSVG). White arrow head points to the AIS of a transfected neuron. Scale bar is 20 μm. This figure was adapted from Yang et al16. (C) Two unhealthy neurons transfected with tdTM and 480 kDa ankyrin-G-GFP were shown. The formation of aggregates (circled in white and enlarged) is a sign of unhealthy neurons. Top: 480 kDa ankyrin-G shows up at the non-AIS region (pointed by white arrow heads. Bottom: Neuron formed 3 AIS and ectopic accumulation of ankyrin-G on soma. Scale bar is 20 μm. Please click here to view a larger version of this figure.
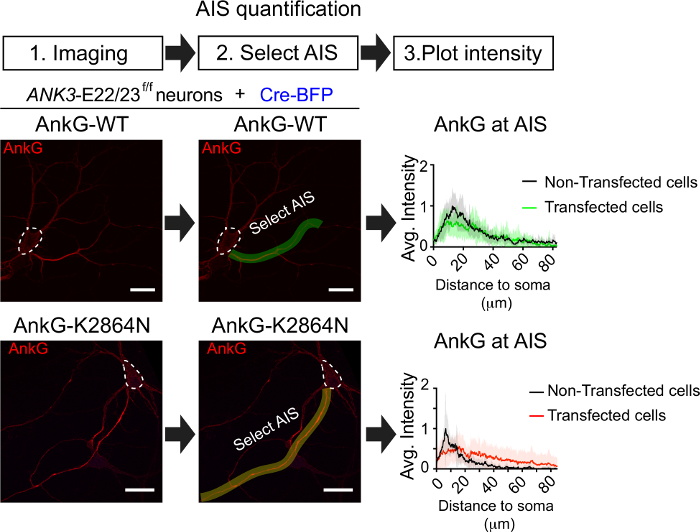
Figure 5: Quantification of AIS structural change. div3 neurons of ANK3-E22/23f/f mice were transfected with Cre-BFP and wild type 480 kDa ankyrin-G or 480 kDa ankyrin-G-K2864N. At 7 div, neurons were fixed and stained for ankyrin-G. Representative images show the ankyrin-G signal at the AIS. The green line and yellow line indicate where the line for AIS intensity measurement was drew. White dash line circled the cell body of the transfected neuron. Scale bar is 20 μm. Average AIS intensity for both conditions is plotted alone distance and aligned with non-transfected cells (n=10). Please click here to view a larger version of this figure.
