Three-Dimensional Motor Nerve Organoid Generation
Summary
This protocol provides a comprehensive procedure to fabricate human iPS cell-derived motor nerve organoid through spontaneous assembly of a robust bundle of axons extended from a spheroid in a tissue culture chip.
Abstract
A fascicle of axons is one of the major structural motifs observed in the nervous system. Disruption of axon fascicles could cause developmental and neurodegenerative diseases. Although numerous studies of axons have been conducted, our understanding of formation and dysfunction of axon fascicles is still limited due to the lack of robust three-dimensional in vitro models. Here, we describe a step-by-step protocol for the rapid generation of a motor nerve organoid (MNO) from human induced pluripotent stem (iPS) cells in a microfluidic-based tissue culture chip. First, fabrication of chips used for the method is described. From human iPS cells, a motor neuron spheroid (MNS) is formed. Next, the differentiated MNS is transferred into the chip. Thereafter, axons spontaneously grow out of the spheroid and assemble into a fascicle within a microchannel equipped in the chip, which generates an MNO tissue carrying a bundle of axons extended from the spheroid. For the downstream analysis, MNOs can be taken out of the chip to be fixed for morphological analyses or dissected for biochemical analyses, as well as calcium imaging and multi-electrode array recordings. MNOs generated with this protocol can facilitate drug testing and screening and can contribute to understanding of mechanisms underlying development and diseases of axon fascicles.
Introduction
Spinal motor neurons (MN) extend axons to skeletal muscles to control body motion. Their axonal trajectories are highly organized and regulated in the developmental process. Despite many studies on axon extension and guidance1, mechanisms for organized axon bundle formation are still under investigation. Axons of motor neurons are often damaged by neurodegenerative diseases such as amyotrophic lateral sclerosis (ALS)2, but pathophysiological mechanisms of the damage on axon fascicles are poorly understood. Thus, a physiological and pathological model to recapitulate axon bundle formation and regression is required in the field.
A human stem cell-derived motor neuron is a promising platform for understanding the development and diseases such as ALS3. Human induced pluripotent stem cells (iPS cells) can be used to model diseases using patient-derived cells. To date, various differentiation methods from pluripotent stem cells into MN have been reported4,5,6. However, axons of neurons in two-dimensional culture are randomly oriented and do not recapitulate in vivo microenvironment within developing nerves in which axons are unidirectionally assembled through dense axo-axonal interactions7. To overcome this issue, we have developed a technique to generate a three-dimensional tissue resembling motor nerve from human iPS cells8, and named the tissue as motor nerve organoid (MNO). The MNO consists of cell bodies located in a motor neuron spheroid (MNS) and an axonal fascicle extended out from the spheroid. The axons in the fascicle are unidirectionally oriented, which resembles axons in developing motor nerves. Hence, MNOs uniquely provide a physiological axonal microenvironment, which was not done by any other previously developed neuronal culture methods.
In this protocol, we describe methods for tissue culture chips fabrication, rapid motor neuron differentiation, and motor nerve organoid formation in developed chips. Our tissue culture chip is very simple, and it only contains a compartment for accepting a spheroid, a microchannel for forming an axon bundle, and a compartment for housing axon terminals. The device does not contain complex structures including microgrooves or micropore filters that are often used to separate axons and cell bodies by size9,10. Hence our devices can be easily fabricated by following the steps described in this protocol if a photolithography setup is available.
Rapid differentiation of human iPS cells is achieved with an optimized combination of inducing and patterning factors (SB431542, LDN-193189, retinoic acid (RA), and smoothened agonist (SAG)) and acceleration factors (SU5402 and DAPT). It has been reported that the combination of SU5402 and DAPT accelerates the differentiation of peripheral neurons and neural crest cells11. In this protocol, we offer three different methods to generate MNOs, so that readers can decide on a method most suited to their needs. We recommend performing differentiation of human iPS cells after forming a spheroid (the 3D method), since the differentiated MNS can be transferred directly into a tissue culture chip. Alternatively, human iPS cells can be differentiated into motor neurons in monolayer (2D) culture, and then created into three-dimensional motor neuron spheroids as we previously reported8. We have updated the protocol, and with the three-dimensional differentiation method described in this protocol, the transition from 2D to 3D can be avoided and MNOs can be obtained with shorter differentiation time, fewer steps, and reduced technical risks without the dissociation step. Commercially available neurons can be also used to generate MNS to reduce the time for differentiation.
To generate an MNO, we cultured an MNS in the tissue culture chip. The axons elongate from the spheroid and extend into the microchannel in which axons gather and align unidirectionally. This facilitates axo-axonal interaction and spontaneous formation of a tightly assembled unidirectional bundle tissue of axons in the microchannel, which is uniquely achieved by this protocol, whereas either spontaneous bundle formation or guided axonal orientation alone can be achieved by other protocols12,13,14. In a typical experiment, few cells migrate out from spheroids to the microchannel and most cells stay nearby spheroids. This method allows axons to be spontaneously separated out from the spheroids without using size-dependent physical barriers (e.g., microgrooves or micropore filters) to separate axons from cell bodies.
The resultant MNO can be subjected to various examinations, including morphological, biochemical, and physical analyses. The cell body and extended axon bundle can be physically isolated by cutting and can be separately analyzed for downstream experiments, e.g., biochemical assays. Biological materials including RNA and protein can be isolated from just a few axon bundles for regular biochemical assays including RT-PCR and western blotting. Here, we describe a protocol for generating motor nerve organoid from iPS cells, which offers an attractive physiological and pathological model to study the mechanism underlying development and disease of axon fascicles.
Protocol
1. SU-8 mold fabrication by photolithography
NOTE: This procedure involves hazardous chemicals. Use fume hood and PPE throughout.
- Clean the silicon wafer (4-inch in diameter, 1-mm thickness, polished) with acetone and blow with nitrogen gas. Then bake at 180 °C for 3 min to dry.
- Dispense 3 mL of SU-8 2100 on to a cleaned wafer.
- Coat the SU-8 uniformly on the wafer using a spin coater at 500 rpm for 10 s and then, sequentially spin at 1500 rpm for 30 s with an acceleration of 300 rpm/s to obtain a 150 µm thick layer of SU-8.
NOTE: Make sure that the silicon wafer is positioned at the center of the spin coater and correctly fixed by vacuum. - Soft bake the wafer on the hot plate at 50 °C for 10 min, at 65 °C for 7 min, and at 95 °C for 45 min.
- Set the photomask (Figure 1) to the mask aligner and expose UV light (365 nm) for 60 s.
NOTE: Exposure time needs to be optimized by an appropriate dose of UV light. - After exposure, bake the wafer at 65 °C for 6 min, and at 95 °C for 13 min on the hot plate.
- Develop the wafer for 10-20 min in SU-8 developer with agitation using an orbital shaker, changing the developing solution once during the process.
NOTE: Extend the developing time when the debris of SU-8 remains. - Rinse the wafer in isopropanol and gently dry the wafer with nitrogen gas.
- Measure the height of the deposited SU-8 by a measuring microscope and make sure it is approximately 150 µm thick. It can be stored indefinitely at room temperature.
2. PDMS microfluidic-based tissue culture chip fabrication
- Fix the SU-8-deposited wafer to a container (e.g., 15 cm plastic Petri dish) by double side tape.
- Blow the dust off the wafer using nitrogen gas.
- To silanize, put the SU-8-deposited wafer in a vacuum chamber together with a small container (e.g., 35 mm dish). Drop 10 µL of (tridecafluoro-1,1,2,2-tetrahydrooctyl)-1-trichlorosilane into the small container. Do not directly apply the (tridecafluoro-1,1,2,2-tetrahydrooctyl)-1-trichlorosilane to SU-8 wafer.
- Close the vacuum chamber tightly and turn on a vacuum pump for at least 2 h.
- Take a plastic cup and pour the silicone elastomer (e.g., Silpot 184 or equivalently Sylgard 184) and the curing agent at a 10:1 weight ratio. Then, mix well using a spatula and degas in the vacuum chamber until bubbles are removed entirely.
- Pour the PDMS mixture into the container with the SU-8 wafer to the desired thickness (3-4 mm) and degas again to remove bubbles.
- Bake the PDMS in an oven at 60 °C for at least 3 h to fully cure the PDMS.
- After cooling, cut off the cured PDMS from the wafer by using a scalpel or a razor blade.
- To create the two chambers of the tissue culture chip, punch two holes where the two compartments are located by using a 1.5 mm diameter biopsy punch.
- To create a medium reservoir, prepare another PDMS mixture (silicone elastomer and the curing agent at a 10:1 weight ratio) and pour it into a new 10 cm Petri dish. Adjust the pouring volume to 5 mm of thickness of PDMS.
- Bake the PDMS in an oven at 60 °C for at least 3 h to fully cure the PDMS.
- After cooling the PDMS, cut off the cured PDMS with a scalpel to get a rectangular ring.
- Bond the bottom layer with the medium reservoir by applying uncured PDMS between them and baking the assembled PDMS layers. This bonded structure results in the PDMS tissue culture chip.
- Clean the PDMS tissue culture chip by a scotch tape to remove dust and small particles from the surface. PDMS tissue culture chips can be stored at room temperature if protected from dust and UV.
3. Preparation of culture
- Culture medium
NOTE: All media listed below should be filtered for sterilization unless otherwise stated. Prepared media may be stored at 4 °C and used within a month.- To prepare mTeSR Plus medium: combine one bottle of 100 mL mTeSR Plus 5x supplement with one bottle of 400 mL of mTeSR Plus Basal Medium.
- To prepare 100 mL of KSR medium: In 85 mL of DMEM/F12, add 15 mL of Knockout serum replacement (KSR, 15%), 1 mL of commercial glutamine supplement (1%) and 1 mL of non-essential amino acid (NEAA, 1%).
- To prepare 100 mL of N2 medium: In 100 mL of Neurobasal medium, add 1 mL of N2 (100x), 1 mL of commercial glutamine supplement, and 1 mL of NEAA.
- To prepare 250 mL of Maturation medium: In 250 mL of Neurobasal medium, add 5 mL of B27 (2%), 2.5 mL of commercial glutamine supplement (1%), and 2.5 mL of Penicillin/Streptomycin (1%).
- Resuspend the compounds (retinoic acid (RA), SB431542, LDN-193189, SU5402, DAPT, SAG, Y-27632) in cell-culture-grade DMSO to the desired concentration. Prepare aliquots and store them at -20 °C for up to 6 months. The following stock solutions are used: 1 mM RA, 10 mM SB431542, 100 μM LDN-193189, 10 mM SU5402, 10 mM DAPT, 1 mM SAG, 10 mM Y-27632.
- Coating
NOTE: To prevent polymerization of basement membrane matrix by heat, avoid repetitive freeze-thaw cycles. Handle all coating procedures with pre-cooled pipette tips and tubes if possible. The basement membrane matrix should be thawed overnight at 4 °C and aliquoted using pre-chilled pipette tips and tubes. The aliquots can be frozen at -20 °C or -80 °C.- Thaw the frozen aliquot at 4°C on ice. The aliquot should be kept cold during the coating procedure. Using a pre-cooled pipette tip, dilute the basement membrane matrix with ice-cold DMEM/F12 at a ratio of 1:40. Unused diluted basement membrane matrix can be stored at 4 °C for 2-3 days given that no polymerization occurred.
- Add 1 mL of the basement membrane matrix/DMEM-F12 solution to coat one well of the 6 well plate.
- Incubate the plate at room temperature for at least an hour, or 4 °C overnight. Coated plates can be stored at 4 °C for a maximum of one week.
4. Maintenance of iPS cells
NOTE: Undifferentiated iPS cells are maintained in mTeSR Plus medium and sub-cultured when confluency of ≥ 90% is observed in a 6 well plate in this protocol. Minor adjustments may be required for iPS cells cultured in other media.
- Prepare basement membrane matrix-coated dishes as previously mentioned in step 3.2.
- Fully aspirate the mTeSR Plus medium. Wash the well once with PBS and add 0.5 mL of passaging reagent (see Table of Materials). Wait a few seconds and aspirate the solution.
- Incubate the plate at 37 °C in the incubator for 5 min or until cells become round.
NOTE: Incubation time might vary between different iPS cell lines and the confluency. Please check periodically under the microscope to determine the dissociation time during the incubation. - Add 1 mL of mTeSR Plus medium and tap the plate for 30-60 s to detach the colonies.
- Gently mix 1 mL of the cell suspension solution with 7 mL of fresh mTeSR plus medium. Do not pipette more than 5 times.
- Plate at a ratio of 1:8. Typically add 1 mL of the suspension from step 4.5 and add 1 mL of mTeSR plus media supplemented with 5-10 μM of Y-27632 (ROCK inhibitor). Passage dilution ratio depends on iPSC line.
- Place the cell in a 5% CO2/37 °C incubator. On the next day, remove Y-27632 by adding fresh mTeSR Plus medium. Thereafter, change media every other day initially, and every day as the cell reaches higher confluency.
5. Differentiation of iPS cells into motor neurons
NOTE: All options below (5.2, 5.3, and 5.4) produce MNOs with > 90% efficiency.
- Passaging iPSC for motor neuron differentiation
NOTE: Differentiation can be successfully conducted in either 3D (5.2) or 2D (5.3) protocols.- Allow undifferentiated iPS cells to grow until it reaches confluency of approximately 80% in mTeSR Plus medium in a 6 well plate.
- Completely aspirate the medium. Immediately wash the well once with sterile PBS and add 0.5 mL of cell dissociation solution to the cells.
- Incubate the plate at 37 °C in the incubator for approximately 2-3 min, or until cells become separated and round but remain attached to the well.
- Add 1 mL of medium and gently pipette up and down a few times using a 5 mL serological pipette. Transfer the cell suspension to a 15 mL tube consisting of 4 mL of the medium.
- Centrifuge at 200 x g for 3 min.
- Carefully aspirate the supernatant, leaving the pellet undisturbed, and resuspend the cells in 1 mL of the medium supplemented with 10 µM of Y-27632.
- Count the cells using a hemocytometer and proceed to either 5.2 (3D differentiation), or 5.3 (2D differentiation).
- Formation of motor neuron spheroid (MNS) in 3D differentiation (“3D protocol”)
NOTE: A complete medium change is done daily from Days 0-12 of differentiation (Figure 2).- Seed the iPS cells from step 5.1.7 to a 96 well U bottom plate at 40,000 cells/well in 100 µL of mTeSR Plus supplemented with 10 µM of Y-27632.
- On the next day, replace each well with 100 µL of the fresh medium.
- On days 0 and 1: Aspirate the culture medium and replace with 100 µL of KSR medium (3.1.2) supplemented with 10 µM SB431542 and 100 nM LDN-193189.
- On days 2 and 3: Aspirate the culture medium and replace with 100 µL of KSR medium supplemented with 10 µM SB431542, 100 nM LDN-193189, 5 µM DAPT, 5 µM SU5402, 1 µM RA and 1 µM SAG.
- On days 4 and 5: Prepare a mixed medium consisting of 75% KSR medium and 25% N2 medium (3.1.3). Then, aspirate the culture medium and replace with 100 µL of mixed medium supplemented with 10 µM SB431542, 100 nM LDN-193189, 5 µM DAPT, 5 µM SU5402, 1 µM RA, and 1 µM SAG.
- On days 6 and 7: Prepare a mixed medium consisting of 50% KSR medium and 50% N2 medium. Then, aspirate culture medium and replace it with 100 µL of mixed medium supplemented with 5 µM DAPT, 5 µM SU5402, 1 µM retinoic acid and 1 µM SAG.
- On days 8 and 9: Prepare a mixed medium consisting of 25% KSR medium and 75% N2 medium. Then, aspirate culture medium and replace with 100 µL of mixed medium supplemented with 5 µM DAPT, 5 µM SU5402, 1 µM RA and 1 µM SAG.
- On days 10 and 11: Replace medium with 100 µl of N2 medium supplemented with 5 µM DAPT, 5 µM SU5402, 1 µM RA and 1 µM SAG.
- On day 12: Proceed to transfer the MNs into the tissue culture chip (Step 6) or replace medium with 100 µL of the Maturation medium (3.1.4) supplemented with 20 ng/mL brain-derived neurotrophic factor (BDNF).
NOTE: The MNS can be transferred to the tissue culture chip starting from day 12 until day 19. Spheroids that are not transferred should be kept cultured in 96 well U bottom plates in Maturation medium supplemented with 20 ng/mL BDNF until transfer.
- (Alternative option) 2D differentiation and transition to 3D MNS (“2D protocol”)
- Aspirate the coating solution from a pre-coated 12 well plate.
- Seed the iPS cells from Step 5.1.7 at a density of 100,000 – 200,000 cells per well in mTeSR Plus medium with 10 µM of Y-27632.
NOTE: Continue culturing the undifferentiated iPS cells in mTeSR Plus medium without Y-27632 until the cells reach 80% confluency if the cells are too sparse for the next step (5.3.3). - On days 0 and 1: Aspirate culture medium and replace with 1 mL of KSR medium (3.1.2) supplemented with 10 µM SB431542 and 100 nM LDN-193189.
- On days 2 and 3: Aspirate culture medium and replace it with 1 mL of KSR medium supplemented with 10 µM SB431542, 100 nM LDN-193189, 5 µM DAPT, 5 µM SU5402, 1 µM RA and 1 µM SAG.
- On days 4 and 5: Prepare a mixed medium consisting of 75% KSR medium and 25% N2 medium (3.1.3). Aspirate culture medium and replace with 1 mL of mixed medium supplemented with 10 µM SB431542, 100 nM LDN-193189, 5 µM DAPT, 5 µM SU5402, 1 µM RA, and 1 µM SAG.
- On days 6 and 7: Prepare a mixed medium consisting of 50% KSR medium and 50% N2 medium. Aspirate the culture medium and replace it with 1 mL of mixed medium supplemented with 5 µM DAPT, 5 µM SU5402, 1 µM retinoic acid and 1 µM SAG.
- On days 8 and 9: Prepare a mixed medium consisting of 25% KSR medium and 75% N2 medium. Aspirate the culture medium and replace with 1 mL of the mixed medium supplemented with 5 µM DAPT, 5 µM SU5402, 1 µM RA and 1 µM SAG.
- On days 10 and 11: Replace medium with 1 mL of N2 medium supplemented with 5 µM DAPT, 5 µM SU5402, 1 µM RA and 1 µM SAG.
- On day 12: Aspirate the differentiation medium, quickly wash well once with PBS and add 0.5 mL of the cell detachment medium. Place the plate in a 37 °C incubator for 1-3 min (e.g., if using TrypLE Express) or 20-30 min (e.g., if using Accutase).
- Using a P1000 pipette, gently collect the cells and transfer the cells into a 15 mL conical tube with fresh Maturation medium and centrifuge at 200 x g for 3 min. If cells are clumpy, gently pipette up and down a few times. Do not pipette too much as this may cause damage to the cells.
- Aspirate the supernatant and resuspend the pellet in 1 mL of Maturation medium (3.1.4) supplemented with 20 ng/mL BDNF.
- Count the cell using a hemocytometer. Plate the cells at 10,000-40,000 cells per well in a 96 well U bottom plate in Maturation medium supplemented with 20 ng/ml of BDNF. Initial seeding density should be optimized depending on iPS cell line and condition of cells so that the diameter of the spheroid is 800-900 μm when introduced into the tissue culture chip. In most cases, start at 20,000 cells per well initially, and then increase or decrease the number of cells according to the size.
- Culture for additional 3-10 days until the cells form a spheroid with a smooth edge.
- (Alternative Option): MNS formation from motor neurons
NOTE: Commercially available human iPS cell-derived motor neurons (see Table of Materials) can be used to generate MNOs instead of differentiating from human iPS cells.- After thawing the cryovial of motor neurons, quickly resuspend the cells with 9 mL of the motor neurons medium. Spin down at 400 x g for 5 min at room temperature.
- Aspirate the supernatant and resuspend the pellet with the motor neuron medium.
- Follow the same steps as above (5.3.12- 5.3.13) to produce MNSs.
6. Preparation of the tissue culture chip for motor nerve organoid (MNO) formation
- Sterilize the prepared PDMS (from step 2.13) by immersing it in 70% ethanol in the Petri dish for at least 1 h.
NOTE: All following steps should be manipulated in a biosafety cabinet. - Sterilize the microscope glass (76 x 52 mm) by immersing it in 70% ethanol in the Petri dish.
- During the drying process of the microscope glass, place the PDMS device on the half wet-microscope glass and let it dry completely by waiting overnight. Once it is completely dried, the PDMS device should adhere to the glass.
NOTE: This bonding is not permanent to allow detaching of the PDMS devices from the microscope glass after the culture for tissue collection. Permanent bonding by oxygen plasma can be used to maximize adhesion between PDMS and glass, but it would prohibit disassembly of the chips and tissue collection. - Coat the surface of the microchannel in PDMS device and microscope glass with 30 μL of diluted basement membrane matrix in DMEM/F12 (1:40) by making a droplet on one side of the inlet of the channel and then aspirate the solution from the other side of the inlet with a pipette or suction pump (Figure 3A). Do not aspirate too much volume of solution to avoid bubble contamination.
- Then, incubate the PDMS device for 1 hour at room temperature or overnight at 4 °C in a secondary container (e.g., Petri dish).
7. Motor nerve organoid (MNO) formation
- Replace the coating solution with pre-warmed 150 µL of Maturation medium supplemented with 20 ng/mL of BDNF just before use.
- Then, place the MNS from step 5.2.9 or 5.3.13 into the inlet of the microchannel using a micropipette with a wide-bore tip. MNS can spontaneously settle down at the bottom of the device by gravity. Do not apply too much pressure when injecting the MNS (Figure 3B).
NOTE: If the MNS is stuck on the sidewall of a hole in a tissue culture chip, gently aspirate the solution from another side of the inlet. - Fill a small reservoir (e.g., a cap of 15 mL tube) with sterile water and place it nearby the tissue culture chip in the secondary container to prevent medium evaporation. Then, place it in a 5% CO2/37 °C incubator.
- For a medium change, aspirate the exhausted culture medium from the center of the medium reservoir (Figure 3C). Do not aspirate all the medium and the tissue.
- Gently add fresh Maturation medium (with BDNF). The medium should be changed every 2-3 days. Do not dry the culture medium at any time during the culture. Axons grow from an MNS into the channel and spontaneously assemble into a single bundle in 2-3 weeks, which result in the formation of an MNO. MNO can be cultured for more than additional 1 month in the device.
8. Downstream analysis of MNO
- Whole-mount immunostaining
- Bring the device or dishes to a chemical fume hood. Fix MNO by adding approximately equal volume of 8% paraformaldehyde (PFA) to the media to achieve a final concentration of 4% PFA. Peel off the PDMS device from the glass and incubate for 15-20 min at room temperature.
- Wash the MNO with PBS and then repeat PBS washing 2x.
- Permeabilize the MNO with 0.2% of Triton X-100 in PBS and incubate for 5 min at room temperature.
- Wash the MNO with PBS and then repeat PBS washing 2x. Then, block the MNO with PBS containing 1% BSA and incubate for 1 h at room temperature.
- Dilute primary antibodies in PBS containing 0.1% BSA and incubate MNO with the primary antibody solution overnight at 4 °C. Wash the MNO with PBS three times.
- Dilute secondary antibodies in PBS with 0.1% BSA and incubate MNO with the secondary antibody solution for 2 h at room temperature. Then wash the MNO three times with PBS.
- Stain the MNO with Hoechst in PBS containing 0.2% Triton-X and incubate for 5 min at room temperature.
- Wash the MNO three times with PBS. The immunostained MNO is ready for imaging with fluorescent or confocal laser microscopes.
- Tissue collection and isolation of axon bundle
- Pour 10 mL of HBSS solution into a 10 cm Petri dish. Then, immerse the entire PDMS device in the HBSS solution.
- Carefully detach the PDMS from the microscope glass under a stereomicroscope. When the tissues stick to the PDMS device, gently apply 1 mL of HBSS solution from the top of the hole by using a pipette.
- Once the tissue comes off from the PDMS, gently apply 1 mL of HBSS on the microscope glass to completely detach the PDMS from the microscope glass.
- To isolate an axon bundle from a spheroid, cut the axon bundle with a surgical knife or tweezer under the microscope. Cut the axon bundle slightly far away (> 1 mm) from the spheroid to avoid contamination of migrated cells.
- Isolated axon bundles and spheroids can be further analyzed by various downstream analyses such as RT-PCR, RNA-seq, and western blotting.
- The PDMS culture chip can be reused. To clean the culture chip, sonicate the culture chip within distilled water for 15 min. Then, sonicate the chip in distilled water supplemented with 1% detergent. Wash the culture chip with distilled water 5 times.
- Calcium imaging
NOTE: Neuronal activity can be measured with calcium indicator both before and after collection of tissue from the tissue culture chip (from 8.2). The protocol is based on a specific commercial kit (see Table of Materials). Alternatively, other Calcium imaging kits or equivalent methods can be used.- Wash the MNO with PBS (without Ca2+ and Mg2+) for three times.
- Incubate the tissue with 5 μM of Fluo-4AM in recording medium (20 mM HEPES, 115 mM NaCl, 5.4 mM KCl, 0.8 mM MgCl2, 1.8 mM, CaCl2, 13.8 mM glucose) for 30-60 min at 37 °C. An addition of 0.01-0.02 % of Pluronic F-127 can assist uptake of Fluo-4 AM into the cells.
- Then, wash the tissue with PBS (without Ca2+ and Mg2+) and replace it with recording medium.
- Acquire time-lapse images by using fluorescent microscopy with a GFP/Cy2 filter set. Set the exposure time less than 20 ms per frame.
- Open the acquired movie file as an image sequence using Image J.
- If using a color CCD or CMOS camera, convert RGB images to 16-bit mono images. Open “Analyze | Tools | ROI Manager”, Draw the region of interest and click “Add”. Click “Multi Measure”. Mean of intensity in multiple ROI will be shown in the Result.
- Plot the change of signal intensity using general data analysis software.
- Measurement of neuronal activity by Multi-electrode array
NOTE: The activity of motor neurons can be captured by a multi-electrode array (MEA).- After creating an MNO, transfer it onto a basement membrane matrix coated MEA probe. Position the tissue on electrodes.
- Add 200 µL of Maturation medium and incubate 1-2 h at 37 °C to allow the MNO to attach to the surface.
- Replace the medium with recording medium (8.3.2). Optionally place mesh and weight on top of an MNO to increase contract with the electrodes.
- Set the MEA probe to the recording head stage. Wipe the contact between the MEA probe and the head stage with 70% ethanol.
- Start recording neuronal activity by following instructions of the manufacturers of the MEA.
Representative Results
Motor neurons were differentiated within 12-14 days in 3D differentiation procedures (Figure 4 and Figure 5). Importantly, more than 60% of cells expressed motor neuron marker HB9 during the differentiation. Immunohistochemistry revealed that approximately 80% of the cells in the MNS were SMI32-positive motor neurons. HB9 and SMI32 are the established early-stage motor neuron markers15,16. The expression of HB9 and SMI32 are the key parameters that need to be confirmed to ensure cellular identity of motor neurons. After the introduction of an MNS into the culture chip, axons extend into the channel and an axon bundle forms. Due to microchannels serving as physical guides, axons elongate from the MNS and form a bundle by axo-axonal interaction (Figure 6A). It is essential to confirm the formation of the axon bundle by microscopic observation to confirm the generation of an MNO. A successful MNO bears an axon bundle wider than 50 μm and few isolated axons out of the bundle in the channel. Initial elongation of axons can be observed 24 hours after the introduction of the spheroid. Within the next 3-4 days, the axons reached to the center of the microchannel and then reaches to the other end within an additional 10 days (Figure 6A). Consequently, the axons assembled and formed a straight and unidirectional bundle in 2-3 weeks in a chip, and neuronal activities were observed thereafter.
Motor nerve organoids can be collected from the chip by detaching the PDMS from the microscope glass for biological analysis (Figure 6B). Axon bundles and cell bodies can be dissected and isolated by cutting using a surgical knife or tweezer under a microscope (Figure 6B). These biological materials including RNA and protein can be used for regular biochemical assays such as RT-PCR and western blotting. In axon bundles of MNOs, nuclear or dendritic maker proteins are not detected in western blotting (Figure 6C).
In combination with a calcium indicator (Fluo-4 AM), neuronal activity can be captured in the tissue culture chip. Spontaneous activity of motor neurons in the spheroid and the axon bundle were observed within the MNO. Also, the neural activities were observed by using a multi-electrode array system.
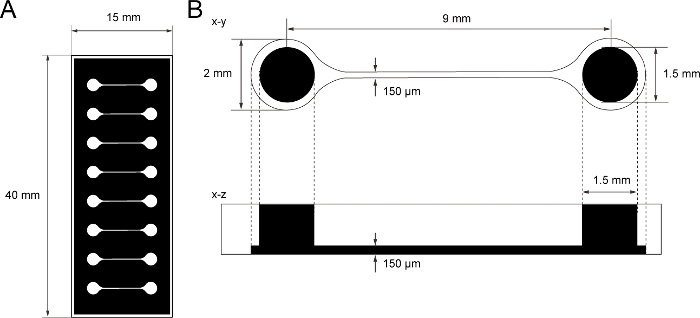
Figure 1: The dimension of PDMS tissue culture chip.
(A) Photomask of the tissue culture chip. (B) Dimensions of microchannel in the tissue culture chip. The diameter of the base chamber for holding motor neuron spheroid is 2 mm and the hole of PDMS above the chamber is 1.5 mm. The width and height of a microchannel bridging two chambers are both 150 μm. Please click here to view a larger version of this figure.
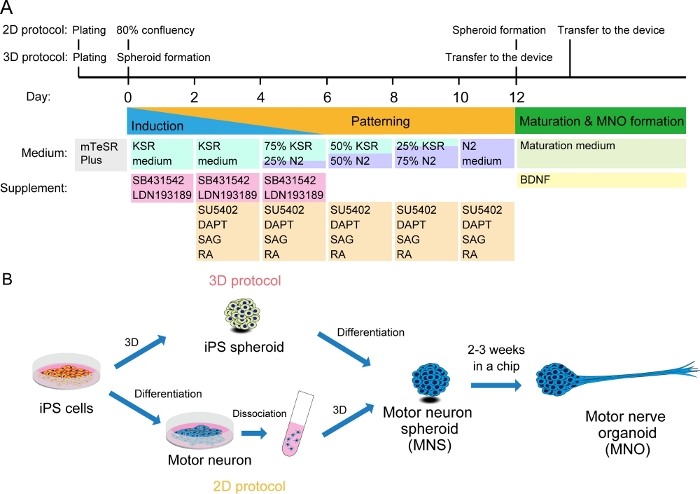
Figure 2: Schematic illustration of motor neuron differentiation.
(A) The differentiation steps involved neural induction, patterning into motor neuron lineage, and maturation of motor neurons. (B) Two options to create motor neuron spheroid (MNS) from iPS cells: 3D protocol, and a 2D protocol with dissociation step of motor neurons. Motor nerve organoid (MNO) can be obtained by both protocols. Please click here to view a larger version of this figure.
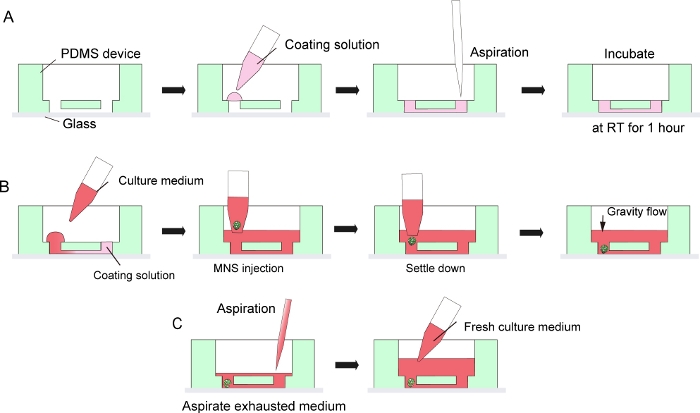
Figure 3: Step by step protocol for basement membrane matrix coating and motor neuron spheroid introduction.
(A) Basement membrane matrix coating in the channel of the tissue culture chip. (B) MNS introduction into the hole of the chip. (C) Culture medium change by aspiration of exhausted medium. Please click here to view a larger version of this figure.
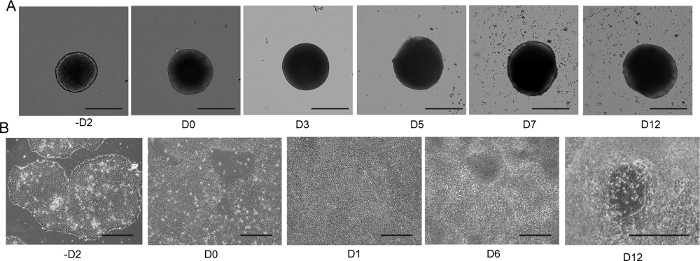
Figure 4: 2D and 3D motor neuron differentiation.
(A) Time course of representative 3D MNS differentiation (3D protocol). The size of the MNS gradually increased over time. Scale bar: 500 μm. (B) Time course of 2D differentiation on -D2, D0, D1, D6, D12 (2D protocol). Scale bar: 500 μm. Please click here to view a larger version of this figure.
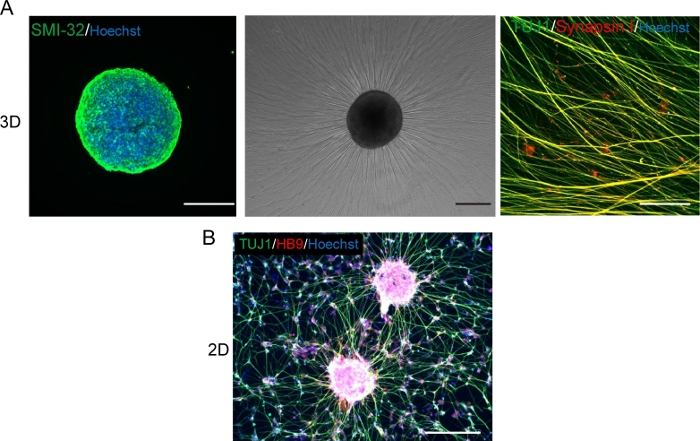
Figure 5: Characterization of motor neurons.
(A) (Left) A cryosection of a MNS stained with SMI-32 antibody and DAPI. (Middle) A phase-contrast image of replated MNS on a basement membrane matrix-coated surface. Axonal elongation was observed. (Right) Axons of replated MNS stained with Synapsin I and Tuj1 antibodies. Scale bar: 500 μm (Left and Middle) and 50μm (Right). (B) A representative image of 2D motor neurons immunostained with Tuj and HB9 antibodies. Scale bar: 200 μm. Please click here to view a larger version of this figure.
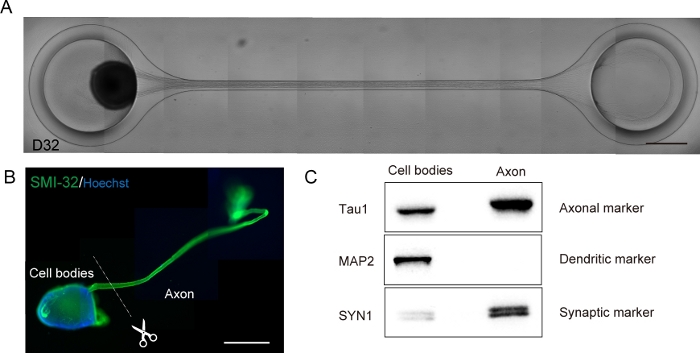
Figure 6: Characterization of a motor nerve organoid (MNO) generated in a culture chip.
(A) Representative images of axon elongation and thick axon bundle formation on D32. Scale bar: 500 μm. (B) Immunostaining of motor nerve organoid (MNO) by SMI-32 and DAPI. Axons and cell bodies can be isolated by physically cutting. Scale bar: 1 mm. (C) Purity of the protein from axons and cell bodies quantified by western blotting. MAP2, a dendritic marker, was not detected in axonal protein, whereas axonal marker Tau1 is enriched in the axonal protein. Please click here to view a larger version of this figure.
Discussion
This protocol describes the formation of a motor nerve organoid (MNO) which has an axon bundle extended from a motor neuron spheroid generated from human iPS cells. The formed axon bundle is thick, flexible, and well-organized in unidirectional structures. By dissecting the axon bundle, high-purity axonal protein and RNA can be obtained sufficiently for biochemical analyses. Neuronal activity can be measured in axon bundles and spheroids with calcium imaging. Contamination of nuclear and dendritic proteins in the axonal lysate was not detected by western blotting, demonstrating that our method efficiently separated axons from cell bodies and dendrites.
One of the advantages of this protocol is the rapid differentiation and generation of MNO equipped with an axon bundle, in which all processes can be done in 4 weeks with the 3D protocol and 5-6 weeks using the 2D protocol. This is short compared to other protocols which typically take 3-4 weeks to simply differentiate into MN from embryonic stem cells and iPS cells17 and it takes an additional 2-4 weeks to obtain axonal elongation. The 3D protocol is generally preferred over the 2D protocol because of the shorter differentiation time, fewer steps, and reduced technical risks without the dissociation step compared to the 2D protocol. The microfluidic-based tissue culture chip was designed in a manner so that the axons of MNS can elongate toward the other compartment through the microchannel, which facilitates formation of a bundle of axons by inducing axo-axonal interactions and affinity between axons. Because of the simple experimental set-up, all the protocols described here can not only be performed by bioengineers, who are familiar with the manipulation of a tissue culture chip, but also biologists and neuroscientists who are not familiar with microfluidics and microfabrication techniques. It should be noted that Steps 1 and 2 can be also performed by using an external fabrication service.
One of the critical steps to accomplish the protocol is a sequential change of culture medium. It is advised to completely change the culture medium at each step during the differentiation so that factors in the spent medium do not disturb the MN differentiation. Another critical point of this protocol is to maintain undifferentiated iPS cells in good quality. Quality of the initial iPS cell culture significantly affects the efficiency to obtain motor neurons and MNO. Another point is that the diameter of MNS should be smaller than the size of the hole of the chip (1.5 mm). Larger spheroids cannot enter the chamber, and potentially experience severe hypoxic necrosis in the center part. The size of MNSs can be controlled by changing an initial seeding number of iPS cells (in 3D protocol) or motor neurons (in 2D protocol). The seeding density of cells should be optimized for each iPS cell line.
Compartmentalized microfluidic devices with microgrooves and small pore filters have been widely used to separate axons from cell bodies and dendrites. This technique can also separate axons from cell bodies and dendrite, with superior abundance of axons in bundled tissues. Compared to the other methods, one major limitation of this method is that it cannot separate two different culture media in the current design of the tissue culture chip, which hinders capability of co-culture of two different cells that require two distinct media. Another limitation is that the PDMS chip put predetermined restriction on size of the tissue. A spheroid larger than the hole cannot enter the chamber, and the axon bundle cannot grow thicker than the width of microfluidic channel.
This method can be applied to other types of neurons. Our group has shown capability to model a cerebral tract by using a modified method combined with cerebral organoid techniques18. Cortical spheroids were introduced into both compartments and the axons spontaneously elongated reciprocally toward each spheroid, and subsequently an axon bundle formed spontaneously. As a result, two cortical spheroids can be connected through an axon bundle, and the tissue could be obtained as one piece. This demonstrates that the approach is highly versatile to form axon bundle tissues regardless of neuronal cell types. In this protocol, human iPS cells were used, however, other stem cells including human ES cells and human neural stem cells may be used with modifications to the presented protocol. 3D spheroids of neurons can be generated by diverse protocols19,20. This method of making tissues with an axon bundle can potentially be combined in the future with the other differentiation protocols for making 3D MN spheroid. In addition, the thickness and the length of the axon bundle can be controlled by simply changing the width and height of microchannels of the tissue culture chip for future developments.
We believe that this protocol can be used for drug testing and screening and can contribute to the understanding of mechanisms underlying the development and diseases of axon fascicles.
Disclosures
The authors have nothing to disclose.
Acknowledgements
This study was supported by the Japan Society for the Promotion of Science (JSPS) Grants-in-Aid for Scientific Research 17H05661 and 18K19903, Core-2-core program, and Beyond AI institute.
Materials
| (Tridecafluoro-1,1,2,2-tetrahydrooctyl)-1-trichlorosilane | Sigma | 440302 | |
| 16% Paraformaldehyde (formaldehyde) aqueous solution | Electron Microscopy Sciences | 15710 | |
| 200µl Wide Bore Pipet Tips | BMBio | BMT-200WRS | |
| 6-well plates | Violamo | 2-8588-01 | |
| Accutase | ICT | AT104 | |
| B-27 Supplement (50X) | Gibco | 17504044 | |
| Bovine serum albumin | Sigma | A6003 | |
| Brain-derived neurotrophic factor (BDNF) | Wako | 020-12913 | |
| CO2 incubator | Panasonic | MCO-18AIC | |
| Cryostor CS10 | Stem Cell Technologies | 07959 | |
| DAPT | Sigma | D5942 | |
| DMEM/F12 | Sigma | D8437 | |
| Fluo-4 AM | Dojindo Laboratories | CS22 | |
| GlutaMAX Supplement | Gibco | 35050-061 | |
| Growth factor reduced Matrigel (basement membrane matrix) | Corning | 354230 | |
| HB9 Antibody | Santa Cruz | sc-22542 | |
| HBSS | Wako | 085-09355 | |
| Hoechst 33342 | Sigma | 14533 | |
| iCell motor neuron (commercially available human iPS cell-derived motor neurons) | Cellular Dynamics | R1051 | |
| Isopropyl alcohol (IPA) | Wako | 166-04836 | |
| Knock Out Serum Replacement | Gibco | 10828028 | |
| LDN193189 | Sigma | SML0559 | |
| MEA probe | Alpha MED Scientific inc | MED-P5004A | |
| MEM Non-essential Amino Acid Solution (100x) (NEAA) | Sigma | M7145 | |
| Microscope Glass | Matsunami | S9111 | |
| mTeSR Plus | Stem Cell Technologies | 05825 | |
| N2 supplement | Wako | 141-08941 | |
| Neurobasal medium | Gibco | 21103049 | |
| Penicillin-streptomycin | Gibco | 15140122 | |
| Photoresist SU-8 2100 | Microchem | #SU-8 2100 | |
| Prime surface 96U | Sumitomo Bakelite | MS-9096U | |
| ReLeSR (passaging reagent) | Stem Cell Technologies | 05872 | |
| Retinoic acid | Wako | 186-01114 | |
| SAG | Sigma | SML1314 | |
| SB431542 | Wako | 192-16541 | |
| Silicon wafer | SUMCO | PW-100-100 | |
| Silpot 184 w/c kit | Dow Toray | Silpot 184 w/c kit | |
| Smi32 Antibody | Biolegend | 801701 | |
| SU5402 | Sigma | SML0443 | |
| SU-8 Developer | Microchem | Y020100 | |
| Synapsin I Antibody | Millipore | Ab1543 | |
| TrypLE Express liquid without phenol red (dissociation solution) | Gibco | 12604-021 | |
| Tuj1 Antibody | Biolegend | 801202 | |
| Y-27632 | Wako | 030-24021 |
References
- Raper, J., Mason, C. Cellular strategies of axonal pathfinding. Cold Spring Harbor Perspective Biology. 2 (9), 001933 (2010).
- Ito, Y., et al. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science. 353 (6299), 603-608 (2016).
- Fujimori, K., et al. Modeling sporadic ALS in iPSC-derived motor neurons identifies a potential therapeutic agent. Nature Medicine. 24 (10), 1579-1589 (2018).
- Chen, H., et al. Modeling ALS with iPSCs Reveals that Mutant SOD1 Misregulates Neurofilament Balance in Motor Neurons. Cell Stem Cell. 14 (6), 796-809 (2014).
- Osaki, T., Uzel, S. G. M., Kamm, R. D. Microphysiological 3D model of amyotrophic lateral sclerosis (ALS) from human iPS-derived muscle cells and optogenetic motor neurons. Science Advances. 4 (10), (2018).
- Imamura, K., et al. The Src/c-Abl pathway is a potential therapeutic target in amyotrophic lateral sclerosis. Science Translational Medicine. 9 (391), (2017).
- Wang, L., Marquardt, T. What axons tell each other: axon-axon signaling in nerve and circuit assembly. Current Opinion in Neurobiology. 23 (6), 974-982 (2013).
- Kawada, J., et al. Generation of a Motor Nerve Organoid with Human Stem Cell-Derived Neurons. Stem Cell Reports. 9 (5), 1441-1449 (2017).
- Nagendran, T., Poole, V., Harris, J., Taylor, A. M. Use of Pre-Assembled Plastic Microfluidic Chips for Compartmentalizing Primary Murine Neurons. JoVE. (141), e58421 (2018).
- Paranjape, S. R., Nagendran, T., Poole, V., Harris, J., Taylor, A. M. Compartmentalization of Human Stem Cell-Derived Neurons within Pre-Assembled Plastic Microfluidic Chips. JoVE. (147), e59250 (2019).
- Chambers, S. M., et al. Combined small-molecule inhibition accelerates developmental timing and converts human pluripotent stem cells into nociceptors. Nature Biotechnology. 30 (7), 715-720 (2012).
- Rimington, R. P., Fleming, J. W., Capel, A. J., Wheeler, P. C., Lewis, M. P. Bioengineered model of the human motor unit with physiologically functional neuromuscular junctions. bioRxiv. , (2020).
- Cullen, D. K., et al. Bundled Three-Dimensional Human Axon Tracts Derived from Brain Organoids. iScience. 21, 57-67 (2019).
- Giandomenico, S. L., et al. Cerebral organoids at the air-liquid interface generate diverse nerve tracts with functional output. Nature Neurosciences. 22 (4), 669-679 (2019).
- Egawa, N., et al. Drug screening for ALS using patient-specific induced pluripotent stem cells. Science Translational Medicine. 4 (145), (2012).
- Sances, S., et al. Modeling ALS with motor neurons derived from human induced pluripotent stem cells. Nature Neurosciences. 19 (4), 542-553 (2016).
- Qu, Q., et al. High-efficiency motor neuron differentiation from human pluripotent stem cells and the function of Islet-1. Nature Communications. 5 (1), 3449 (2014).
- Kirihara, T., et al. A Human Induced Pluripotent Stem Cell-Derived Tissue Model of a Cerebral Tract Connecting Two Cortical Regions. iScience. 14, 301-311 (2019).
- Rigamonti, A., et al. Large-Scale Production of Mature Neurons from Human Pluripotent Stem Cells in a Three-Dimensional Suspension Culture System. Stem Cell Reports. 6 (6), 993-1008 (2016).
- Yan, Y., Song, L., Madinya, J., Ma, T., Li, Y. Derivation of Cortical Spheroids from Human Induced Pluripotent Stem Cells in a Suspension Bioreactor. Tissue Engineering Part A. 24 (5-6), 418-431 (2017).
