Various transgenic, viral vector, or inorganic dye-labeling approaches can be used to specifically visualize several retinal cells types and structures in vivo using a simple adaptation of a basic multiphoton microscope. To visualize RGCs and amacrine cells, VGlut2-Cre and VGat-Cre transgenic mice, respectively, were given an intravitreal injection of a Cre-dependent AAV expression construct encoding Twitch2b, a cytoplasmic fluorescence resonance energy transfer (FRET)-based Ca2+ sensor that contains cyan and yellow fluorescent proteins (CFP and YFP, respectively) and the Ca2+ binding domain of troponin15. In VGlut2-Cre mice, RGC somas are clearly discernable, and fascicles of axons are often apparent (Figure 3).
It should be noted that the trajectory of axons and the negative image of the vasculature makes it very straightforward to identify the optic nerve head in VGlut2-Cre mice, which is useful as a landmark in chronic imaging experiments (Figure 3). Although amacrine cells appear less bright than RGCs, possibly due to their smaller soma size and/or less efficient AAV transduction, their somas are still readily apparent in the inner nuclear layer. In contrast to RGCs, amacrine cell neurites are more often observed in the inner plexiform layers (Figure 4). Retinal microglia can be imaged in the Cx3cr1-GFP transgenic mouse line6. Microglia associate with the vasculature, making it possible to find the same region in time-lapse imaging experiments.
This approach can be used to track the dynamics of fine microglia processes, a procedure that has better spatial resolution in single-plane images, or if maximum-intensity projections are prepared focusing on individual cells (Figure 5). The poor axial resolution caused by optical aberration through the mouse lens precludes examination of fine details in the z-dimension. To determine whether this imaging technique can observe degenerative changes in cellular ultrastructure, 1 µL of 50 mM N-methyl-D-aspartate (NMDA) was injected into the vitreous to induce excitotoxic lesion. One day after injection, the microglia demonstrated short processes or amoeboid morphology (Figure 5) in accordance with previous reports16. It should be noted that cells in the Cx3cr1-GFP transgenic line exhibited more uniform and complete fluorescent protein expression across the cellular cohort than in experiments with AAV-mediated delivery of fluorescent protein expression cassettes. The benefits of varied and sparse versus complete and uniform labeling should be considered when designing experiments.
To label retinal vasculature as previously described8, mice were injected intraperitoneally with 200 µL of Evans blue dye (20 mg/mL in sterile saline) 30-60 min before imaging. This led to strong labeling of blood vessels emanating from the optic nerve head (Figure 6). Surprisingly, the fluorescent signal of a single injection persisted for at least seven days. Two distinct methods were used to estimate the dimensions of the in vivo images. First, the same retinal regions were imaged in vivo and after fixation in flattened retinal wholemounts using confocal microscopy (Figure 7). Random cell pairs were selected from four different in vivo samples, and the true distance between cell pairs was measured in confocal scans and matched with in vivo pixel distance to obtain an average pixel size of 0.99 µm with 1x digital magnification. Using similar methods correlating in vivo images with confocal wholemount scans has revealed that a single head position allows for imaging over a roughly 650 mm2 patch of retina.
Repositioning of the head holder along one torsional axis can allow access to a linear region of the retina 2.2 mm in length (not shown). Further, 1 or 2 µm diameter fluorescent microspheres were injected into the eyes of mice and their diameter was measured as full-width half-maximum of line scans from in vivo images with 10x digital zoom. This gave a slightly larger pixel size estimate, but with more variance (Figure 7). Overall, confocal imaging of wholemount samples after completion of in vivo experiments is the most consistent method to assign scale to individual images, as variance in corneal and lens properties may alter image scale from sample to sample.
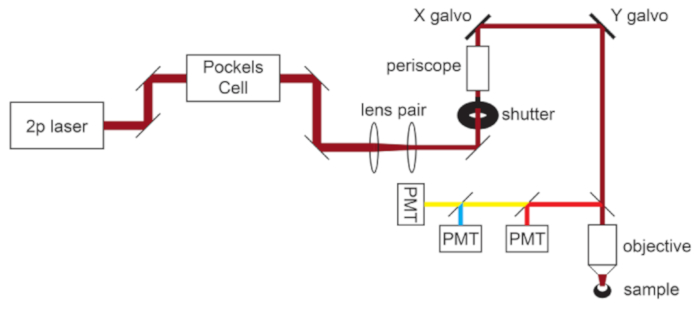
Figure 1: Light path schematic. The basic components of the two-photon microscope used in this protocol consist of a Pockels Cell to modulate laser power, a lens pair to reduce the laser beam diameter to match the back aperture of the microscope objective, and a pair of galvo scan mirrors for beam steering. A pair of steering mirrors is present before each major optical component. The focus is controlled by a motor that drives the objective mount. The emission light path can be customized for different fluorophores by changing out dichroic and barrier filters. A general setup for cyan/yellow/red imaging is displayed in which a short pass dichroic mirror directs red light to the first PMT, and a long pass dichroic mirror paired with appropriate band pass filters is used to separate cyan and yellow emissions. Abbreviation: PMT = photomultiplier tube. Please click here to view a larger version of this figure.

Figure 2: Positioning mice for in vivo imaging. To position the mouse with the pupil on axis with the light path, the anesthetized mouse is first restrained in a head holder, the head is rotated and angled, a large drop of lubricant eye gel is placed on the eye, and the mouse is placed on the stage. A coverslip is mounted in the coverslip holder perpendicular to the light path and lowered down towards the eye. The coverslip should not contact the cornea or mouse head (left), which will be evident if the coverslip is deflected. However, the coverslip should also be close enough to avoid waisting of the droplet (right), because this will have a demagnifying effect on the sample. After applying the gel immersion and securing the coverslip, the stage should be moved in place directly under the microscope objective. Please click here to view a larger version of this figure.
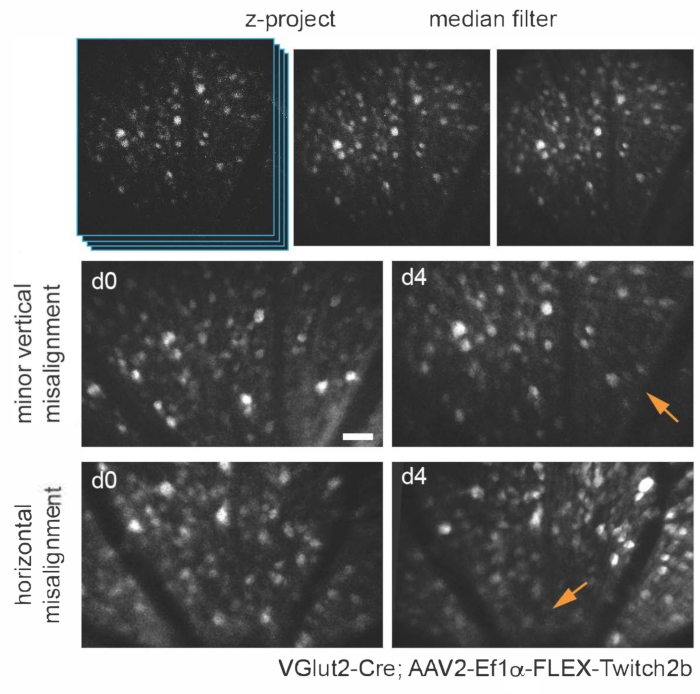
Figure 3: Imaging retinal ganglion cells. For image display, maximum intensity projections with the z-planes containing cells of interest are created, and resultant images are median-filtered to remove PMT shot noise. Two examples of retinal ganglion cells labelled by injecting AAV-EF1α-FLEX-Twitch2b into VGlut2-Cre transgenic mice are shown, specifically the CFP signal. Images were acquired at sessions four days apart, and vascular landmarks were used to return to the same region near the optic nerve head. The optic nerve head is oriented towards the bottom of the image. Although both samples show some variance in orientation (regions with decreased intensity are indicated with arrows), most cells are present at both time points. Scale bar = approximately 50 µm. Abbreviations: PMT = photomultiplier tube; AAV = adeno-associated virus; EF1α = elongation factor-1alpha; FLEX = flip-excision; VGlut2 = vesicular glutamate transporter 2; CFP = cyan fluorescent protein. Please click here to view a larger version of this figure.
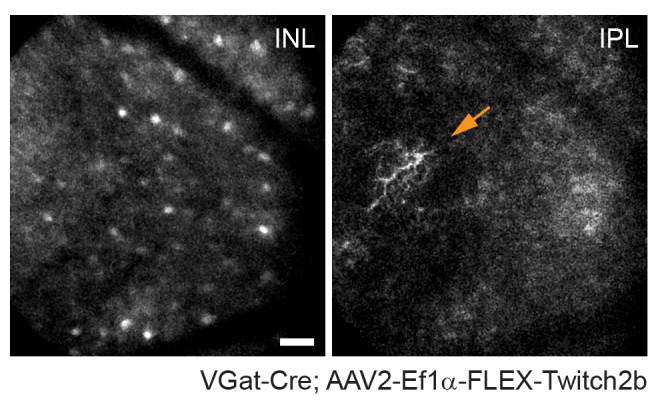
Figure 4: Imaging amacrine cells. Amacrine cells were labeled by injecting AAV-EF1α-FLEX-Twitch2b into VGat-Cre transgenic mice. The CFP signal of Twitch 2b is specifically shown. Small maximum-intensity projections focused on the depths of the inner nuclear layer indicate amacrine cell somas, while focusing on the inner plexiform resolves amacrine cell neurites (arrow). The optic nerve head is oriented towards the right of the image. Scale bar = approximately 50 µm. Abbreviations: AAV = adeno-associated virus; EF1α = elongation factor-1alpha; FLEX = flip-excision; VGat = vesicular gamma aminobutyric acid transporter; CFP = cyan fluorescent protein; INL = inner nuclear layer; IPL = inner plexiform layer. Please click here to view a larger version of this figure.
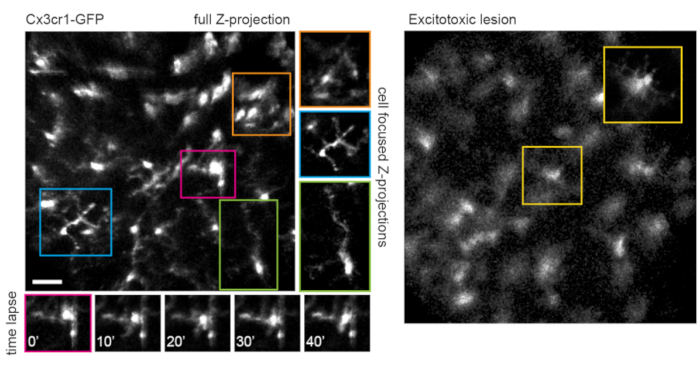
Figure 5: Imaging microglia. The transgenic mouse line Cx3cr1-GFP was used to label microglia. A maximum-intensity projection of the full scan volume shows many microglia, some with fine process detail that can be resolved. Note that cells towards the lower left of the field have less distortion in the maximum projection than those towards the upper right due to parallax in this region. Maximum-intensity projections containing only the cell of interest significantly reduces this parallax (center, boxed in corresponding colors). Furthermore, this imaging strategy can document the dynamics of fine microglia process remodeling (lower panels). Comparatively, many microglia can be seen with short processes or amoeboid morphology one day after an excitotoxic lesion by intravitreal injection of 50 mM NMDA (right). Scale bar = approximately 50 µm. Abbreviations: GFP = green fluorescent protein; Cx3cr1 = Cx3 chemokine receptor 1; NMDA = N-methyl-D-aspartate. Please click here to view a larger version of this figure.
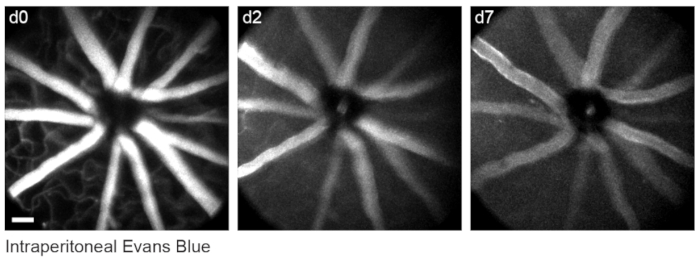
Figure 6: Labelling vascular landmarks. Mice were injected with 200 µL of 20 mg/mL Evans blue intraperitoneally 30-60 min prior to the first imaging session. Full thickness maximum-intensity projections demonstrate lasting fluorescence in the retinal vasculature that persisted for at least seven days. Scale bar = approximately 50 µm. Please click here to view a larger version of this figure.
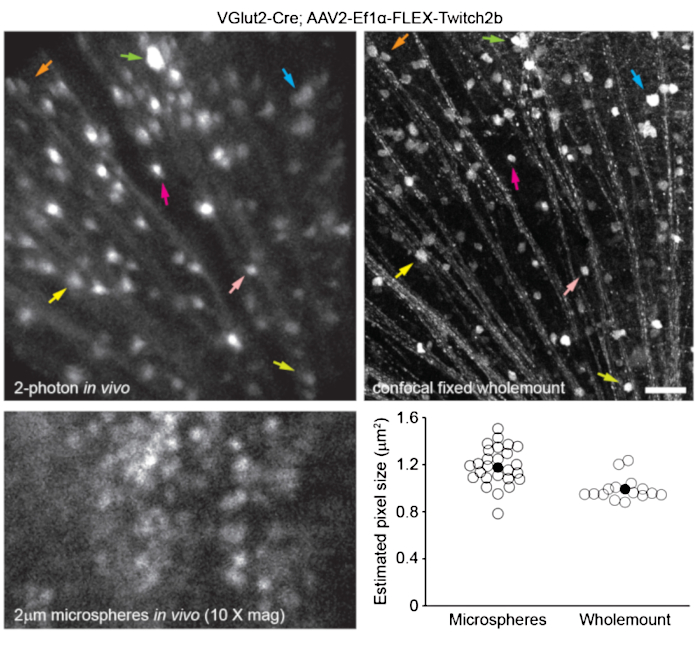
Figure 7: Image dimensions. Retinal ganglion cells labelled by injecting AAV-EF1α-FLEX-Twitch2b into VGlut2-Cre transgenic mice were imaged in vivo, and the same region was then imaged by confocal laser scanning microscopy after fixation and wholemount preparation of the retina. Yellow fluorescent protein channel is shown for both. Colored arrow pairs indicate the same cell in both preparations (upper panels). Single-plane image of 2 µm diameter fluorescent microspheres injected intravitreally and imaged in vivo (lower left panel). Microspheres did not settle and thus were in constant motion making measurement of axial resolution impossible. Pixel sizes calculated from full-width half-maximum measurements of in vivo fluorescent microspheres or correlative confocal measurements taken from 2-4 retinas per group (lower right). Scale bar = 50 µm. Abbreviations: AAV = adeno-associated virus; EF1α = elongation factor-1alpha; FLEX = flip-excision; VGlut2 = vesicular glutamate transporter 2. Please click here to view a larger version of this figure.
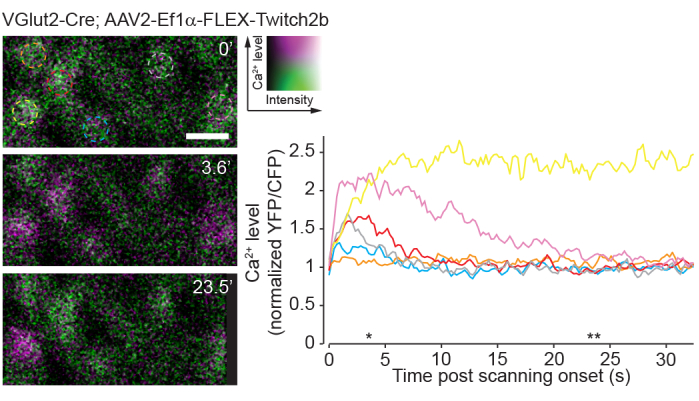
Figure 8: Calcium activity induced by two-photon scanning. Retinal ganglion cells labelled by injecting AAV-EF1α-FLEX-Twitch2b into VGlut2-Cre transgenic mice, YFP is pseudocolored magenta and CFP green, imaged in a single plane as a time series at 4.22 Hz. All RGCs had a similar starting YFP/CFP ratio. Most responded with an increase in FRET ratio (excluding the orange cell), and one maintained a high YFP/CFP ratio throughout the time series (yellow cell). YFP/CFP ratios were normalized to the first frame average, and colored circles match with colored traces. Asterisks indicate time points with representative images displayed on left. Scale bar = 20 µm. Abbreviations: AAV = adeno-associated virus; EF1α = elongation factor-1alpha; FLEX = flip-excision; VGlut2 = vesicular glutamate transporter 2; YFP = yellow fluorescent protein; CFP = cyan fluorescent protein; RGCs = retinal ganglion cells; FRET = fluorescence resonance energy transfer. Please click here to view a larger version of this figure.
