In vitro Modeling for Neurological Diseases using Direct Conversion from Fibroblasts to Neuronal Progenitor Cells and Differentiation into Astrocytes
Summary
We describe a protocol to reprogram human skin-derived fibroblasts into induced Neuronal Progenitor Cells (iNPCs), and their subsequent differentiation into induced Astrocytes (iAs). This method leads to fast and reproducible generation of iNPCs and iAs in large quantities.
Abstract
Research on neurological disorders focuses primarily on the impact of neurons on disease mechanisms. Limited availability of animal models severely impacts the study of cell type specific contributions to disease. Moreover, animal models usually do not reflect variability in mutations and disease courses seen in human patients. Reprogramming methods for generation of induced pluripotent stem cells (iPSCs) have revolutionized patient specific research and created valuable tools for studying disease mechanisms. However, iPSC technology has disadvantages such as time, labor commitment, clonal selectivity and loss of epigenetic markers. Recent modifications of these methods allow more direct generation of cell lineages or specific cell types, bypassing clonal isolation or a pluripotent stem cell state. We have developed a rapid direct conversion method to generate induced Neuronal Progenitor Cells (iNPCs) from skin fibroblasts utilizing retroviral vectors in combination with neuralizing media. The iNPCs can be differentiated into neurons (iNs) oligodendrocytes (iOs) and astrocytes (iAs). iAs production facilitates rapid drug and disease mechanism testing as differentiation from iNPCs only takes 5 days. Moreover, iAs are easy to work with and are generated in pure populations at large numbers. We developed a highly reproducible co-culture assay using mouse GFP+ neurons and patient derived iAs to evaluate potential therapeutic strategies for numerous neurological and neurodegenerative disorders. Importantly, the iA assays are scalable to 384-well format facilitating the evaluation of multiple small molecules in one plate. This approach allows simultaneous therapeutic evaluation of multiple patient cell lines with diverse genetic background. Easy production and storage of iAs and capacity to screen multiple compounds in one assay renders this methodology adaptable for personalized medicine.
Introduction
Understanding underlying disease mechanisms is critical for neurological diseases as it aids in the development of potential therapeutic strategies. While animal models have historically been the gold standard for researching diseases of the nervous system, the direct translation of potential therapies into a clinical setting often shows limited success1,2,3. Reasons for the lack of translation are lack of variability of genetic background and mutations in mice, incomplete display of disease phenotypes and variation in drug sensitivity or dosing in human patients that are not pictured well in inbred mouse strains. Furthermore, for many rare neurological disorders, no or few animal models are available. Studying disease mechanisms directly in relevant human cell types could facilitate research and improve translation to the clinic. For CNS disorders, primary human cells are difficult to retrieve and represent a very limited source as biopsies are invasive or can only be retrieved post-mortem at the end-stage of the disease. In the past 15 years, the development of cellular reprogramming technology has rapidly expanded the ability to model human neurological and neurodegenerative diseases in vitro.
Traditional methods of cell reprogramming involve reprogramming fibroblasts or other cell types into induced pluripotent stem cells (iPSCs) capable of differentiating into cell types from all three germ layers4. Takahashi and Yamanaka were the first to show that re-expression of four stem cell transcription factors is sufficient to redirect somatic cells to become iPSCs5. iPSCs can then be further differentiated into specific cell types. However, the drawback of this process is that it is labor intensive and time consuming to generate and isolate stable stem cell clones. Somatic mutations or specific aberrancies of the mother cell are also maintained in the stem cell clone and can impact results of the study6. In addition, increasing evidence suggests that the reprogramming process to stem cells erases valuable epigenetic changes that could influence cellular disease mechanisms4,7,8. In recent years, researchers focused on modified reprogramming methods allowing a more direct generation of different cell types while bypassing the pluripotent stem cell state9,10,11,12 (reviewed in 13,14). Initial breakthroughs revealed in vitro combinatorial reprogramming of fibroblasts to cardiomyocytes15, neurons16 and hepatocytes17 by ectopic expression of multiple lineage-specific transcription factors or microRNAs18. This was followed by studies directly reprograming cells to model neurological disorders19,20. As mentioned above, such direct reprogramming or conversion protocols have potential advantages over classical iPSC protocols including speed and the ablation of the clonal isolation step. Moreover, evidence suggests that these protocols allow to maintain a larger amount of epigenetic information related to the age of the patient and likely the same holds true for disease relevant markers7,8.
To date, most in vitro research on neurological disorders has been focused primarily on neurons. However, it is well known that other cell types such as astrocytes, microglia and oligodendrocytes play a critical role in disease pathology and progression of neurodegenerative disorders such as Alzheimer's disease (AD), Amyotrophic Lateral Sclerosis (ALS), Parkinson's disease (PD), Huntington's Disease (HD), Multiple Sclerosis (MS), and other neurological pathologies such as Rett syndrome (RTT), sleep disorders, addiction, epilepsy, lysosomal storage disorders, depression, migraine and pathological pain21,22,23,24,25,26. Astrocytes are the most abundant cell type in the central nervous system (CNS) and until recently, they have been broadly neglected from a pathological point of view7. They are now known to have a critical role in supporting almost all homeostatic functions of the healthy CNS. Additionally, it is apparent that they have an important pathological impact and are very valuable in understanding disease mechanism and evaluating therapeutic strategies19.
In the current description, we detail a protocol for direct conversion of human patient fibroblasts into induced Neuronal Progenitor Cells (iNPCs) using retroviral vectors expressing the Yamanaka reprogramming factors (Klf4, Oct3/4, Sox2 and c-Myc) and subsequent exposure to neuralizing media. Previous studies have used different combinations of transcriptional factors for direct reprogramming to neural cells (reviewed in 14), but the factors used in the described protocol are broadly available either as plasmids for in house production of viral vectors, or as commercially available ready to go viral reprogramming kits. The resulting iNPCs can be further differentiated into induced neurons (iNs)27, oligodendrocytes (iOs)24 and astrocytes (iAS)19 to study their role in neurological diseases. Our protocol does not involve time-consuming generation of iPSCs which most available protocols utilize for derivation of human astrocytes28. Approximately 98% to 100% of directly reprogrammed iNPCs can differentiate into GFAP-positive astrocytes29 as compared to only 2% using direct conversion method from fibroblasts to astrocytes30. Recently, a comparative transcriptomic study using our described reprogramming method has shown that iNPCs reprogrammed from donor fibroblasts can retain ageing features at transcriptional and functional level similar to age matched postmortem astrocytes and primary astrocytes29. Thus, this conversion protocol is a powerful tool to investigate disease mechanisms and evaluate novel therapeutic approaches for age-related neurodegenerative and neurological disorders.
Here we describe the protocol utilized to generate iNPCs and further differentiation into iAs, which are the most suited for larger scale small molecular screening assays. Disease mechanisms and drug testing with iAs can be done using different methodologies such as co-cultures with neurons or metabolic and biochemical analysis. The advantage of this system is the speed and ease of maintenance of these cell lines compared to iPSCs. Moreover, iNPCs can be frozen in small portions for iAs differentiation which facilitates drug testing under constant conditions over extended periods of time. Overall, comparison of larger sample numbers becomes possible this way which opens the door to evaluate therapeutic potential of compounds in a larger and more representative patient population.
Protocol
| Media | Reagent | Amount to mix | Final concentration (%) |
| Fibroblast media | DMEM, high glucose, GlutaMAX | 500 mL | 89 |
| Fetal Bovine Serum | 50 mL | 10 | |
| Antibiotic-Antimycotic | 5 mL | 1 | |
| Base media | DMEM/F12 + Glutamax | 500 mL | 97 |
| N-2 | 5 mL | 1 | |
| B-27 | 5 mL | 1 | |
| Antibiotic-Antimycotic | 5 mL | 1 | |
| Conversion media | Base media | 50 mL | 99.9 |
| FGF | 1 µL | 0.02 (20 ng/mL) | |
| EGF | 1 µL | 0.02 (20 ng/mL) | |
| Heparin | 50 µL | 0.1 (5 µg/mL) | |
| Neural Progenitor Cell (NPC) Media | DMEM/F12 + Glutamax | 500 mL | 96.9 |
| N-2 | 5 mL | 1 | |
| B-27 | 5 mL | 1 | |
| Antibiotic-Antimycotic | 5 mL | 1 | |
| FGF | 10 uL | 0.002 | |
| Astrocyte Media | DMEM, high glucose, GlutaMAX | 500 mL | 89 |
| Fetal Bovine Serum | 50 mL | 10 | |
| Antibiotic-Antimycotic | 5 mL | 1 | |
| N-2 | 1 mL | 0.2 | |
| Media must be filtered after mixing all components. Conversion media (with factors) must be prepared fresh every week. | |||
Table 1. Media recipes for all cell types included in the protocol. Media should be filtered after mixing all components with either a sterile vacuum filtration system for big volumes or syringe and 0.2 um syringe filters. Conversion media (with factors) must be prepared fresh every week.
1. Direct conversion of adult human fibroblasts to neuronal progenitor cells
NOTE: A schematic timeline of this protocol can be reviewed in Meyer (2014)19.
- Use primary human skin fibroblasts that can be obtained from cell banks or skin biopsies31. Culture cells at 37 °C in a 5% CO2 tissue culture incubator. Passage cells for at least 1-2 passages post thawing prior to use in the experiment. Once the cells are ready, coat a well of a 6-well plate with human fibronectin (1:200 diluted in PBS) at room temperature for 15-20 min.
- When the cells are ready to be plated, remove the fibronectin from the dish and immediately add cell solution or 2 mL of fibroblast media (Table 1) – do not let the dish dry out prior to adding media. Depending on how fast the cells grow, plate 150,000 (fast growing) – 200,000 (slow growing) fibroblasts in two wells of a human fibronectin coated 6-well plate each.
NOTE: The cells should be around 70% confluent for viral transduction to be able to keep them in the same plate for several more days after transduction. It can be beneficial to seed at two different densities in order to have a choice the next day for conversion. - The day after seeding, check the fibroblasts under the microscope and verify cell density (between 70-85%) and even seeding throughout the well for optimal results.
- Transduce one well with all four retroviral vectors (Oct3/4, Klf4, Sox2 and c-Myc) – use a multiplicity of infection (MOI) of 10 for fast growing or 15 for slow growing fibroblasts (transduction efficiency should exceed 70% or more positive cells for each vector). Add regular fibroblast medium to viral mix to a total volume of 1 mL per well and incubate overnight at 37 °C in a 5% CO2 tissue culture incubator. Leave the second well untreated and return cells to tissue culture incubator.
NOTE: Retroviral vectors can be purchased by a vendor or made in-house, protocols are available online32. - The day after transduction, wash cells 1x with PBS and add 1 mL of fresh fibroblast medium.
- The next day, change medium to conversion medium. Wash cells 1x with PBS to get rid of the residual fibroblast medium, and then add 1 mL of conversion medium. Change the media of the untreated well to conversion media as well.
NOTE: Some morphological changes can be observed even by changing the media on the fibroblasts that did not receive retroviral vector treatment, thus having an untreated well (optional) will be helpful when determining effects of the viral vectors. Cells should start changing morphology 2-4 days after switching to conversion media. - Observe cells under the microscope and watch for changes in morphology (Figure 1). Cell media should be replaced daily throughout the conversion process (1-2 mL of conversion media, Table 1) and for subsequent iNPC culture. Change medium by carefully removing 70% of medium from the well while making sure that the cells remain covered in the remaining 30% of media.
NOTE: Media with growth factors in it needs to be consumed within 1 week of preparation.- Continue to observe cells and change media daily (1-2 mL of conversion media).
- Some cell lines start forming loose elevated round formations growing in ball like structures or loose neuronal spheres (Supplemental Figure 1 and 19,33). Take care to not detach these cells. If the balls detach, they can be collected and re-plated in a new fibronectin coated well of a 6-well plate.
NOTE: If they are expanded on their own, the cells will have reduced diversity compared to the initial plate and may represent a distinctly different cell line. We recommend combining these cells with the rest of the converted cells at the next round of splitting.
- After about 5-6 days in conversion media (up to 12 days for difficult cell lines) or when cells become very dense, passage them 1:2 or maximum 1:3.
NOTE: It is very important to keep the cells at a high density throughout the conversion process.
2. Conversion and iNPC splitting procedure
- Warm up cell detachment solution (e.g., Accutase) and coat an appropriate number of wells of a 6-well with fibronectin (1:200 in PBS, 1 mL of coating volume) for 15-20 min at room temperature. Fibronectin coating can be longer without negative impact, but care should be taken to not shorten the coating time.
- Wash cells carefully with PBS without detaching any cells (use the wall of the well to apply PBS gently to the cells). Carefully remove PBS with pipettes or by aspiration.
- Add 0.5 mL of cell detachment solution and incubate 2-3 min at 37 °C in a tissue culture incubator. Check under the microscope to verify that most cells have detached. If all cells have come off, add 2 mL of fresh conversion media and gently pipette up and down 2-3 times to dissociate clumps (if required).
- Collect the cells in a 15 mL tube and add additional 3 mL of media to further dilute the cell detachment solution. Wash the well with the extra media first to ensure all cells have been collected.
- Centrifuge for 4 min at 200 x g at room temperature.
NOTE: Converting cells or iNPCs are extremely sensitive to the presence of cell detachment solution in the media. It is absolutely necessary to remove the residual enzyme by centrifugation and withdrawal of supernatant. - Remove supernatant from cell pellet and resuspend the cells in 1 mL of fresh medium. Gently pipette up and down 2-3 times to resolve cell clumps. Remove fibronectin from coated 6-wells and add 1 mL of fresh media to each well immediately (do not let fibronectin dry). For a split ratio of 1:2, add 0.5 mL of the cell suspension in one well and the other 0.5 mL in a second well.
- Culture cells at 37 °C in a tissue culture incubator.
- Distribute the cells by gently shaking the 6-well in north-south-east-west direction (not circular) and put in tissue culture incubator.
- Observe the cells under the microscope the next day and change media (1-2 mL). Change media every day until the cells are ready to be split again.
NOTE: At this point, the cell proliferation should start to accelerate, and cells should be ready for a second split within 2-3 days (4 days for very slow growing lines).- At this new split, seed one well of cells in conversion media and the other well in NPC media containing only FGF at increased concentration without EGF/heparin (Table 1).
NOTE: Some cell lines reach a stagnant state in conversion media after about 10 days and switching to NPC media might help with cell growth. In addition, NPCs have a more specific, smaller shape, when grown in NPC media19. At this point, the iNPCs are ready for differentiation and further use.
- At this new split, seed one well of cells in conversion media and the other well in NPC media containing only FGF at increased concentration without EGF/heparin (Table 1).
- Keep changing media (1-2 mL) of the iNPCs daily.
- Expand NPCs into 10 cm dishes by combining two or three confluent wells of a 6-well. The media volume for 10 cm dishes is typically 12-15 mL.
- At the next split, further expand these cells to three or four 10 cm dishes (12-15 mL of media) for generations of large numbers of cell stocks. 10 cm dishes are usually split at 1:3 or 1:4 every 3-4 days depending on proliferation rate.
3. Generating induced astrocytes from NPCs
- To make astrocytes from fresh iNPCs, seed iNPCs (in 10 cm fibronectin coated plates) directly in 10 mL astrocyte medium (Table 1) so that they are around 10% confluency the following day. Culture cells at 37 °C in a 5% CO2 tissue culture incubator.
NOTE: The recommended seeding density is usually 50 µL of the cell resuspension of a 10 cm dish of NPCs, provided they are diluted to a final volume based on their split ratio (e.g., 3 mL for a 1:3 split, 4 mL for a 1:4 split, etc.). - Change medium (10 mL astrocyte media) of the iAs three days after plating. Keep the cells differentiating for 5-7 days.
NOTE: iAs do not tolerate multiple passages well, therefore it is better to make fresh astrocytes every time you split the NPCs. - To seed for experimentation, split the astrocytes using trypsin or cell detachment solution as described in steps 2.1-2.7. Recommended seeding densities are included in Table 2.
| Plate Type | Astrocytes per Well |
| 384-well | 2500 |
| 96-well | 10000 |
| 24-well | 40000 |
| 6-well | 150000 |
Table 2. Recommended seeding density of iAs per plate area. Typical number of iAs seeded to generate a monolayer on the most common tissue culture plates.
4. Preparing and defrosting portions for astrocyte differentiation from frozen NPC stocks (alternative to step 3)
NOTE: As an alternative to maintaining iNPCs in culture, astrocytes can also be produced directly from a frozen stock. To do so, the iNPCs are frozen in smaller portions. Table 3 shows suggested freezing and thawing volumes for portions to be defrosted into a 10 cm dish containing 10 mL of Astrocyte media.
| Portion Size | Cell Suspension (µL) | Freezing media (µL) | Total volume (µL) | Defrosts Into |
| 4x | 400 | 400 | 800 | Two 10-cm dishes |
| 2x | 200 | 200 | 400 | One 10-cm dish |
Table 3. Instructions on how to portion iNPC to generate iAs. Proportion of iNPC suspension and freezing media to generate portions. Note that the final DMSO concentration is 10% when cell suspension is mixed with freezing media.
- To make astrocytes from frozen iNPC stocks, remove portion stock vial from liquid nitrogen storage tank and quickly thaw at 37 °C. As soon as cells are defrosted, pipette cell solution in a 15 mL tube containing 5 mL astrocyte media.
- Centrifuge for 4 min at 200 x g and room temperature. Remove the supernatant and resuspend in 1 mL of fresh astrocyte medium.
- Add 9 mL of fresh astrocyte medium to a fibronectin coated 10 cm dish and add 1 mL of cell solution. Gently distributing cells in north-south-east-west motion. Culture cells at 37 °C in a 5% CO2 tissue culture incubator.
Representative Results
This protocol allows the rapid and easy generation of iNPCs directly from human skin fibroblasts using retroviruses containing the Yamanaka factors. This method allows bypassing the stem cell state and the need for clonal selection, thereby avoiding clonal variation. Important steps to keep in mind while cells are undergoing the conversion process are the fibroblast seeding density, media pH and keeping the cells at an optimal confluency. Examples of optimal splitting confluency and morphology changes during the conversion process can be found in Figure 1.
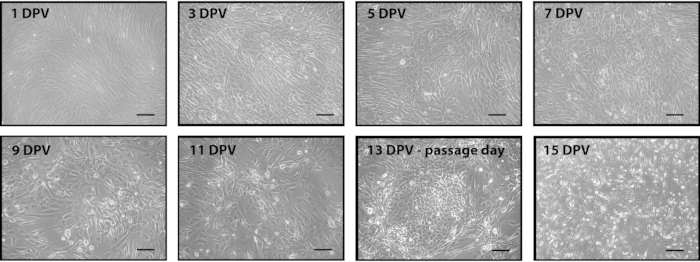
Figure 1. Representative images of the conversion process after adding the retroviral mix of Yamanaka factors. Fibroblasts were seeded and transduced twenty-four hours later with Yamanaka factors. Two days post virus (DPV), media was changed to conversion media. Cells were cultured in conversion media until they were ready to passage (13 DPV). Cells were seeded post passage to reach 80% confluence the following day (14 DPV). Subsequent passages maintained this density until neuronal progenitor cells begin rapidly dividing. Scale bar is 200 µm. Please click here to view a larger version of this figure.
After the conversion process is complete, the NPCs will show strongly reduced expression or no expression of fibroblast markers and morphology, and express NPC specific cell markers (Figure 2). Moreover, they can also be used for generating different cell lines, like iNs, iOs, and iAs.
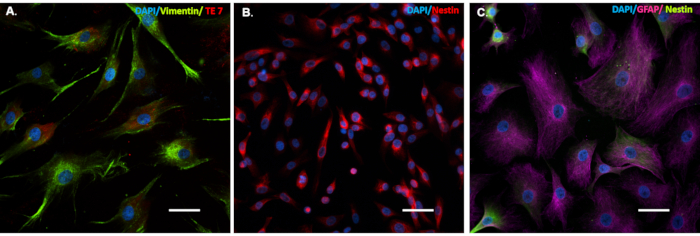
Figure 2. Cells that undergo the conversion process express cell-specific markers. Patient fibroblasts (A), induced Neuronal Progenitor Cells (iNPCs (B) and induced Astrocytes (iAs (C) cells were seeded on glass coverslips in a 24 well tissue culture treated plate. Cells were immunostained for: fibroblast specific cell markers (A), Vimentin (green) and TE7 (red), iNPC specific cell marker (B), Nestin (red), and iAs cell marker (C) GFAP (purple) and iNPC marker Nestin (green). Scale bar is 50 µm. Please click here to view a larger version of this figure.
In our experience, iAs in particular are very valuable for drug and disease mechanism testing as they can be generated in pure populations (98% or more GFAP positive cells29) at reproducible large numbers. iAs can be differentiated from iNPCs by taking a small aliquot of cells during splitting or a previously frozen iNPC portion and directly plating in astrocyte media. Important considerations during this process are maintaining the iNPCs initial seeding density low (Figure 3 and Figure 4), as a high density has been shown to hinder the differentiation process (Figure 4) and paying additional attention to the media pH, as acidification can activate even healthy astrocytes.
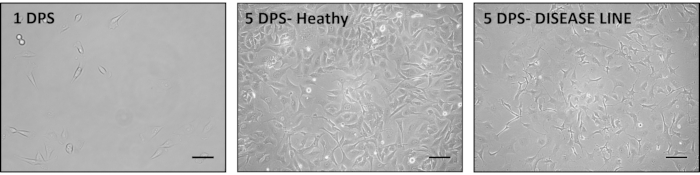
Figure 3. Representative images of the iAs generation process from iNPCs. iNPCs are seeded in Astrocyte media (Table 1) at a low seeding density. A good seeding density is about 10% on the first day post seeding (DPS) (left); however, this density can be adjusted according to the growth rate of cells. Typical iAs morphology can be observed after 5 DPS (middle). In some cases, aberrant astrocyte morphology can be observed, with long, spiky extensions. This change is indicative of astrocyte activation and can be secondary to disease state or incorrect culturing techniques (right). Scale bar is 200 µm. Please click here to view a larger version of this figure.
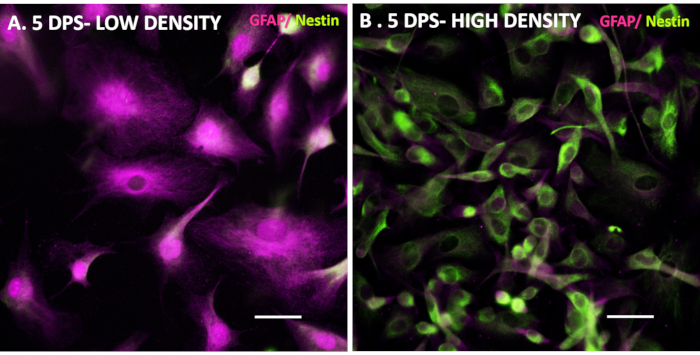
Figure 4. iNPC seeding density affects the iAs differentiation efficiency. Control iNPC lines were seeded at low (A) and high (B) density in Astrocyte media to demonstrate effects of seeding density on differentiation. 5 DPS, the low-density line expressed astrocyte-specific markers, while the high-density line shows a mixture of iNPC and iAs markers with a marked iNPC-like morphology. Scale bar is 50 µm. Please click here to view a larger version of this figure.
Supplemental Figure 1. Additional figures of the conversion process after adding the retroviral mix of Yamanaka factors. Images of the conversion process at 12 DPV (1 day before passage) and 19 DPV (6 days after passage). Note the difference on morphology between cells before and after passage, at 12 DPV cells have a ball-like structure morphology. After a passage and several days on conversion media (table 1) cells start displaying a NPC-like morphology. Scale bar is 200 µm. Please click here to download this File.
Discussion
In summary, the direct conversion is a fast and easy method to generate induced neuronal progenitor cells from patient skin fibroblasts. This method is advantageous because of its speed as well as the large number of cells generated that are easy to maintain. Moreover, increasing evidence suggests that direct reprogramming methods remove less epigenetic marks compared to classic iPSC reprogramming technology, therefore leaving the disease environment more intact4,7,8. The differentiation of iNPCs to astrocytes is very straight forward and highly reproducible even between multiple independent laboratories19,29,33. iNPCs can be frozen in small portions and thawed directly for differentiation purposes, which helps reduce variation between experiments since passage numbers can be kept similar. Astrocytes play a crucial role in disease progression in many neurological diseases and are therefore an interesting cell type to work with. The astrocytes can be used for mechanistic studies, metabolic studies or drug testing assays and are usually grown as monolayers. Moreover, astrocytes can be combined in co-culture systems with mouse or human neurons to assess the impact of astrocytes on neuronal survival and morphology, both of which are good readouts for drug testing34.
In order to successfully use this protocol, a few points and potential limitations need to be kept in mind. The human skin fibroblasts for example vary greatly in their cell division rate as well as response to viral vectors. Since these are not standardized cell lines, they are as variable as mankind itself, each line is different. As for many reprogramming methods, it is easier to reprogram fibroblasts that have a moderate to fast cell division rate versus cells that are barely replicating. The replication rate can be impacted by the skin biopsy quality and processing itself, by the passage number of the fibroblasts as well as handling. When working with primary skin fibroblasts, it is important to not let the cells get too dense to avoid induction of senescence. If the fibroblasts do not grow well, it is best to try a new stock or a lower passage, although in some cases, the disease itself could also play a role. Cell division can sometimes be improved by using 15% or 20% FBS in the fibroblast media compared to the standard 10%.
The quality of the reprogramming viral vectors is of high importance for success. In our hands, retroviral vectors were more efficient compared to lentiviral vectors or other non-integrating viruses; however, this could be dependent on the viral vector quality and purity. Commercial retroviral vector kits usually specify the viral vector concentration and often also a multiplicity of infection for a certain cell line. It is important to keep in mind that primary fibroblasts differ from the cell line used by the companies to determine viral vector uptake. Furthermore, even when ordering the same commercial kit, variation between batches is common. Moreover, the uptake of viral vectors also varies between fibroblast cell lines. Some lines could be more sensitive and therefore transduced cells could die, while other cell lines might be more resistant to viral uptake. Thus, determining transduction efficiency for a given cell line can be important especially if the cell line is growing slowly or previous conversion attempts failed. In our hands, the best way to evaluate how much of a specific retroviral vector is needed for conversion is to perform an immunofluorescent staining with several dilutions of the viral vector. The number of cells expressing the transgene for each vector can then be counted, with the aim of a transduction efficiency of 70% or higher. In our experience, similar MOIs can be used for most cell lines however the MOI can be adjusted if necessary.
During the conversion process, it is critical to not split the cells too early. The time until the cells are ready to be split depends on multiple factors including the primary cell line and its characteristics, the quality of the viral vector used and the media quality. Importantly, the success rate of the conversion is much higher when the cells are kept at high density. Even if the cells start to change morphology, it is better to leave the cells in the first well longer than splitting them prematurely. If the split occurs too early the conversion can halt in its progress and lead the cells to differentiate back to fibroblast-like shape and behavior. Furthermore, diluting them too early can result in more elongated cell bodies compared to the small and compact cell bodies of the NPCs observed previously. Thus, the first splits should always be conservative; it is better to keep the cells too dense with a 1:1 or 1:2 split compared to diluting them too much. Some cell death is expected following the initial split. Of note, the cells always look worse (flatter) the first day after splitting, which is normal. Best judgments on cell shape should be made 2-3 days later. Importantly, the quality of the growth factors and media components is crucial as well. It is important to prepare small amounts of media containing growth factors so that the fully prepared media will only be used for 5-7 days maximum. Do not keep the media at room temperature or 37°C for extended periods of time. Use quickly and refrigerate as soon as the media change is performed. In addition, keeping the volume of media low at 1 mL instead of 2 mL can help especially for cell lines that are more resistant to converting. If the cells consume the media rapidly, which can be observed by a change in media color form red to yellow (acidification rate), adjust the volume of the media to up to 2 mL. Rapid media consumption is a positive sign and often observed at later stages of conversion alongside increased proliferation.
NPCs are also very sensitive to the presence of accutase in the media and it is very important to keep the accutase incubation time short. We recommend an incubation period of 2-4 minutes, check cells frequently under the microscope to see when they have detached. It is essential to centrifuge the cells after splitting to remove the enzyme mix from the media. It is also important to keep the passage number in mind as cell lines can change upon extended culturing leading to alteration of expression profiles. Thus, it is important to freeze many cell stocks at early passages. Cells should be frozen in 10% DMSO and 90% NPC media and stored long term in liquid nitrogen. Due to the lack of serum in this media it is critical to ensure a slow freeze using freezing chambers and once frozen overnight, immediately transfer to liquid nitrogen. While no clonal selection is performed in this protocol, it is possible that extended culturing and passaging leads to natural selection of an initial cell population with favorable growth and survival rate. Therefore, it is important to keep passage number and handling in mind when performing experiments. We prefer to use lower passages for experiments, if possible, we do not exceed passaging the cell lines for more than 20 times. Since NPCs grow rapidly, large numbers of stocks can be frozen at lower passages for experiments. Cell lines with particularly high growth rates are at higher risk of media acidification. Thus, as cell lines grow with different speed, the amount of media might need to be adjusted based on proliferation. If the cells are continuously left in highly acidified media, it can influence the health of both control and disease cell lines. This causes even healthy astrocytes to become reactive, affecting their use in further differentiation and experiments.
For astrocyte differentiation, it is important to keep the cells at low density as high density will prevent the cells from differentiating and they will keep proliferating at higher rates. Thus, seeding density needs to be adjusted for each cell line dependent on their proliferation rate. We recommend trying different seeding densities and perform stainings/RT-PCR with astrocyte markers to ensure proper differentiation. IAs quality can be assessed alongside healthy controls using co-culture assays with neurons. Provided disease pathology does not lead to a reactive phenotype, neurons should remain viable in contact with iAs for several days. Astrocyte morphology can vary greatly between individual cell lines and can be impacted by the disease phenotype as well. Moreover, in diseases where astrocytes are heavily impacted, they might show a reactive phenotype with large, elongated extensions.
Disclosures
The authors have nothing to disclose.
Acknowledgements
We thank all patients and healthy volunteers for donating critical samples to research studies. Moreover, we thank Rochelle Rodrigo for continuous expert technical assistance in tissue culture and Laura Ferraiuolo for independently confirming the technique in her lab as a collaborator. This work received funding from US National Institutes of Health Grants R01 NS644912-4 and RC2 NS69476-01 (to A.L.S.) and Packard Center for ALS Research Grant P2ALS and the Helping Link Foundation. K.M. also received funding from the Swiss National Science Foundation as well as the Young Investigator Development Award from the Muscular Dystrophy Association, The Rett Syndrome Research Trust and the Families of SCN2A foundations.
Materials
| 100mm x 2mm Style dish, Cell culture treated, Nonpyrogenic | Corning | 430167 | Tissue culture |
| 15 ml conical screw cap centrifuge tubes, copolymer polypropylene | USA Scientific | 1475-1611 | Used for lifting and centrifuge cells |
| 50mL Conical Centrifuge Tubes | USA Scientific | 1500-1811 | Media preparation |
| Antibiotic-Antimycotic (100X) | Gibco | 15240062 | Antibiotic with antifungal activity for media preparation |
| B-27 Supplement (50X), serum free | Invitrogen | 17504044 | For NPC and base media |
| Cryogenic vials 1.2ML | Corning | CLS430658-500EA | For freezing cell stocks |
| DMEM, high glucose, GlutaMAX Supplement, pyruvate | Gibco | 10569010 | For fibroblast and Astrocyte media |
| DMEM/F-12, GlutaMAX supplement | Gibco | 10565042 | For NPC and base media |
| DMSO | Sigma | D2438-50ML | For freezing cell stocks |
| Dulbecco’s Phosphate Buffered Saline (PBS) | Gibco | 14190136 | Referred in protocol as PBS. For fibronectin dilution and cell wash |
| EZ Retrovirus iPSC Reprogramming Kit | ALSTEM | RF101 | Retrovirus containing the Yamanaka factors. Virus can also be made in-house. |
| Fetal Bovine Serum, certified | Gibco | 16000-036 | Referred in protocol as FBS . For Fibroblast and Astrocyte media |
| Fisherbrand Sterile Syringes for Single Use | Fisher | 14-955-461 | Filter media (50mL) |
| Heparin sodium salt from porcine intestinal mucosa | Sigma | H3149-10KU | Referred in protocol as Heparin. Used in conversion media. Powder diluted in ultrapure water. Final stock concentration of 5000X |
| Human Plasma Fibronectin Purified Protein | Millipore Sigma | FC010-10MG | Referred in protocol as Fibronectin, used in 1:200 dilution for coating. |
| Isopropanol (technical grade) | Fisher Scientific | S25372A | For freezing cell stocks |
| Mr. Frosty | Thermo Fisher | 5100-0001 | For freezing cell stocks |
| N-2 Supplement (100X) | Gibco | 17502048 | For Astrocyte, NPC and base media |
| Recombinant Human EGF | Preprotech | AF-100-15 | Referred in protocol as EGF, used in conversion media. Powder diluted in PBS, final concentration of 1mg/mL, stored in small frozen aliquots |
| Recombinant Human FGF-basic | Preprotech | 100-18B | Referred in protocol as FGF, used in NPC and conversion media. Powder diluted in PBS, final concentration of 1mg/mL, stored in small frozen aliquots |
| StemPro Accutase Cell Dissociation Reagent | Gibco | A1110501 | Referred in protocol as Accutase. Used for lifting during the conversion process and NPCs. |
| Stericup Quick Release-GP Sterile Vacuum Filtration System | Millipore Sigma | S2GPU05RE | Media filtration |
| Tissue culture plate, 6 well, Flat bottom with Low evaporation lid | Fisher | 08-772-1B | Tissue culture |
| Trypsin-EDTA (0.05%), phenol red | Invitrogen | 25300062 | Lifting of Fibroblasts and Astrocytes |
| Whatman Puradisc 25 syringe filters, 0.2 μm, PES sterile | Millipore Sigma | 80-2502 | Filter media (50mL) |
References
- Leenaars, C. H. C., et al. Animal to human translation: a systematic scoping review of reported concordance rates. Journal of Translational Medicine. 17 (1), 223 (2019).
- Rhrissorrakrai, K., et al. Understanding the limits of animal models as predictors of human biology: lessons learned from the sbv IMPROVER Species Translation Challenge. Bioinformatics. 31 (4), 471-483 (2015).
- Hartung, T. Thoughts on limitations of animal models. Parkinsonism & Related Disorders. 14, 81-83 (2008).
- Ladewig, J., Koch, P., Brustle, O. Leveling Waddington: the emergence of direct programming and the loss of cell fate hierarchies. Nature Reviews Molecular Cell Biology. 14 (4), 225-236 (2013).
- Takahashi, K., Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 126 (4), 663-676 (2006).
- Hamada, M., Malureanu, L. A., Wijshake, T., Zhou, W., van Deursen, J. M. Reprogramming to pluripotency can conceal somatic cell chromosomal instability. PLoS Genetics. 8 (8), 1002913 (2012).
- Stricker, S. H., Gotz, M. Epigenetic regulation of neural lineage elaboration: Implications for therapeutic reprogramming. Neurobiology of Disease. 148, 105174 (2020).
- Tang, Y., Liu, M. L., Zang, T., Zhang, C. L. Direct Reprogramming Rather than iPSC-Based Reprogramming Maintains Aging Hallmarks in Human Motor Neurons. Frontiers in Molecular Neuroscience. 10, 359 (2017).
- Kelaini, S., Cochrane, A., Margariti, A. Direct reprogramming of adult cells: avoiding the pluripotent state. Stem Cells Cloning. 7, 19-29 (2014).
- Kim, E., Tae, G. Direct reprogramming and biomaterials for controlling cell fate. Biomaterials Research. 20, 39 (2016).
- Kim, J., et al. Direct reprogramming of mouse fibroblasts to neural progenitors. Proceedings of the National Academy of Sciences of the United States of America. 108 (19), 7838-7843 (2011).
- Tian, C., et al. Direct conversion of dermal fibroblasts into neural progenitor cells by a novel cocktail of defined factors. Current Molecular Medicine. 12 (2), 126-137 (2012).
- Mertens, J., Marchetto, M. C., Bardy, C., Gage, F. H. Evaluating cell reprogramming, differentiation and conversion technologies in neuroscience. Nature Reviews Neuroscience. 17 (7), 424-437 (2016).
- Kim, J., Ambasudhan, R., Ding, S. Direct lineage reprogramming to neural cells. Current Opinion in Neurobiology. 22 (5), 778-784 (2012).
- Ieda, M., et al. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell. 142 (3), 375-386 (2010).
- Ambasudhan, R., et al. Direct reprogramming of adult human fibroblasts to functional neurons under defined conditions. Cell Stem Cell. 9 (2), 113-118 (2011).
- Kogiso, T., Nagahara, H., Otsuka, M., Shiratori, K., Dowdy, S. F. Transdifferentiation of human fibroblasts into hepatocyte-like cells by defined transcriptional factors. Hepatology International. 7 (3), 937-944 (2013).
- Grath, A., Dai, G. Direct cell reprogramming for tissue engineering and regenerative medicine. Journal of Biological Engineering. 13, 14 (2019).
- Meyer, K., et al. Direct conversion of patient fibroblasts demonstrates non-cell autonomous toxicity of astrocytes to motor neurons in familial and sporadic ALS. Proceedings of the National Academy of Sciences of the United States of America. 111 (2), 829-832 (2014).
- Lee, M., et al. Direct Reprogramming to Human Induced Neuronal Progenitors from Fibroblasts of Familial and Sporadic Parkinson’s Disease Patients. International Journal of Stem Cells. 12 (3), 474-483 (2019).
- Meyer, K., Kaspar, B. K. Glia-neuron interactions in neurological diseases: Testing non-cell autonomy in a dish. Brain Research. 1656, 27-39 (2017).
- Verkhratsky, A., Parpura, V., Pekna, M., Pekny, M., Sofroniew, M. Glia in the pathogenesis of neurodegenerative diseases. Biochemical Society Transactions. 42 (5), 1291-1301 (2014).
- Ettle, B., Schlachetzki, J. C. M., Winkler, J. Oligodendroglia and Myelin in Neurodegenerative Diseases: More Than Just Bystanders. Molecular Neurobiology. 53 (5), 3046-3062 (2016).
- Ferraiuolo, L., et al. Oligodendrocytes contribute to motor neuron death in ALS via SOD1-dependent mechanism. Proceedings of the National Academy of Sciences of the United States of America. 113 (42), 6496-6505 (2016).
- Palpagama, T. H., Waldvogel, H. J., Faull, R. L. M., Kwakowsky, A. The Role of Microglia and Astrocytes in Huntington’s Disease. Frontiers in Molecular Neuroscience. 12, 258 (2019).
- Maezawa, I., Swanberg, S., Harvey, D., LaSalle, J. M., Jin, L. W. Rett syndrome astrocytes are abnormal and spread MeCP2 deficiency through gap junctions. Journal of Neuroscience. 29 (16), 5051-5061 (2009).
- Hautbergue, G. M., et al. SRSF1-dependent nuclear export inhibition of C9ORF72 repeat transcripts prevents neurodegeneration and associated motor deficits. Nature Communications. 8, 16063 (2017).
- Almad, A., Maragakis, N. J. A stocked toolbox for understanding the role of astrocytes in disease. Nature Reviews Neurology. 14 (6), 351-362 (2018).
- Gatto, N., et al. Directly converted astrocytes retain the ageing features of the donor fibroblasts and elucidate the astrocytic contribution to human CNS health and disease. Aging Cell. , (2020).
- Caiazzo, M., et al. Direct conversion of fibroblasts into functional astrocytes by defined transcription factors. Stem Cell Reports. 4 (1), 25-36 (2015).
- Vangipuram, M., Ting, D., Kim, S., Diaz, R., Schule, B. Skin punch biopsy explant culture for derivation of primary human fibroblasts. Journal of Visualized Experiments. (77), e3779 (2013).
- Pear, W. Transient transfection methods for preparation of high-titer retroviral supernatants. Current Protocols in Molecular Biology. , 11 (2001).
- Boczonadi, V., et al. Mutations in glycyl-tRNA synthetase impair mitochondrial metabolism in neurons. Human Molecular Genetics. 27 (12), 2187-2204 (2018).
- Rinaldi, F., Motti, D., Ferraiuolo, L., Kaspar, B. K. High content analysis in amyotrophic lateral sclerosis. Molecular and Cellular Neuroscience. 80, 180-191 (2017).
