Restriction endonucleases (REases) are enzymes that effect sequence-specific double-strand breaks in DNA. The discovery of REases in the 1970s led to the development of recombinant DNA technology, and these enzymes are now indispensable laboratory tools for genetic modification and manipulation1. Type II REases are the most widely used enzymes in this class as they cleave DNA at a fixed location either within or near their recognition sequence. However, there is a great deal of variation among the Type II REases, and they are divided into several subtypes based on particular enzymatic properties rather than being classified according to their evolutionary relationships. Among each subtype, there are frequent exceptions to the classification scheme, and many enzymes belong to multiple subtypes2. Thousands of Type II REases have been identified, and hundreds of them are commercially available.
However, in spite of the diversity among the Type II REases, very few REases have been studied in detail. According to REBASE, the restriction enzyme database established by Sir Richard Roberts in 19753, published kinetics data are available for fewer than 20 of these enzymes. Furthermore, while some REases have been directly observed at the single-molecule level while diffusing along the DNA prior to encountering and binding to their recognition sequence4,5,6,7, there are very few single-molecule studies of their cleavage reaction kinetics. The existing studies either do not report adequate statistics to undertake detailed analysis of the variation in the times at which single cleavage events take place8,9,10 or are not capable of capturing the full distribution of cleavage times11. This type of analysis can reveal the presence of relatively long-lived kinetic intermediates and could lead to better understanding of the mechanisms of REase-mediated DNA cleavage.
At the single-molecule level, biochemical processes are stochastic-the waiting time for a single instance of the process to occur, τ, is variable. However, many measurements of τ can be expected to obey a probability distribution, p(τ), that is indicative of the type of process taking place. For instance, a single-step process, such as the release of a product molecule from an enzyme, will obey Poisson statistics, and p(τ) will take the form of a negative exponential distribution:
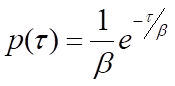
where β is the mean waiting time. Note that the rate of the process, k, will be equal to 1/β, the inverse of the mean waiting time. For processes that require more than one step, p(τ) will be the convolution of the single-exponential distributions for each of the individual steps. A general solution for the convolution of N single-exponential decay functions with identical mean waiting times, β, is the gamma probability distribution:
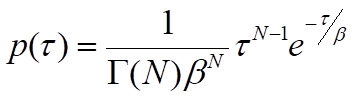
where Γ(N) is the gamma function, which describes the interpolation of the factorial of N-1 to non-integer values of N. Although this general solution can be used as an approximation when the mean waiting times of individual steps are similar, it must be understood that the presence of relatively fast steps will be masked by steps with significantly longer waiting times. In other words, the value of N represents a lower limit on the number of steps12. With an adequate number of waiting-time measurements, the parameters β and N can be estimated by binning the events and fitting the gamma distribution to the resulting histogram or by using a maximum-likelihood estimation approach. This type of analysis can therefore reveal the presence of kinetic steps that cannot be easily resolved in ensemble assays and requires a large number of observations to estimate parameters accurately12,13.
This paper describes a method to use quantum-dot-labeled DNA and total internal reflection fluorescence (TIRF) microscopy to observe hundreds of individual REase-mediated DNA cleavage events in parallel. The design of the assay makes it possible to pool the results of several experiments and can create dwell-time distributions containing thousands of events. The high photostability and brightness of quantum dots permit a 10 Hz time resolution without sacrificing the ability to observe cleavage events occurring even many minutes after the start of the experiment. Good temporal resolution and a broad dynamic range, combined with the ability to collect a large data set, allow accurate characterization of the dwell-time distributions to uncover the presence of multiple kinetic steps in the cleavage pathways of REases, which have turnover rates in the 1 min-1 range. In the case of EcoRV, three kinetic steps can be resolved, all of which have been identified through other means, confirming that the assay is sensitive to the presence of such steps.
Duplex DNA substrates containing the recognition sequence of interest are produced by annealing a biotinylated oligonucleotide to a complementary strand labeled with a single, covalently attached semiconductor nanocrystal quantum dot. These substrates are introduced into a flow channel built on top of a glass coverslip with a lawn of high molecular weight polyethylene glycol (PEG) molecules covalently attached to its surface. The DNA substrates are captured via a biotin-streptavidin-biotin linkage by a fraction of the PEG molecules that have a biotin at their free end. In TIRF microscopy, an evanescent wave that decays exponentially with distance from the glass-liquid interface provides illumination; the penetration depth is on the order of the wavelength of the light used. Under these conditions, only quantum dots that are tethered to the surface by a DNA molecule that has been captured on the functionalized glass surface will be excited. Quantum dots that are free in solution will not be constrained within the illuminated region and therefore will not luminesce. If the DNA tethering a quantum dot to the surface is cleaved, that quantum dot will be free to diffuse away from the surface, and it will disappear from the fluorescence image.
Although many Type II REases are known to bind DNA in the absence of magnesium14, all require magnesium to mediate DNA cleavage15. These REases can bind the surface-immobilized DNA in the absence of magnesium. When magnesium-containing buffer is flowed through a channel with REase prebound to the DNA, cleavage begins immediately, as indicated by the disappearance of quantum dots. The synchronization achieved by prebinding the REase molecules, and then initiating DNA cleavage by introducing magnesium, facilitates measurement of the lag time to the completion of DNA cleavage independently for each molecule in the population of enzymes observed in an experiment. Fluorescein is included as a tracer dye in the magnesium-containing buffer to indicate the arrival of magnesium into the field of view. As no enzyme is included in the magnesium-containing buffer, the lag time from the arrival of magnesium-containing buffer to the disappearance of each quantum dot indicates the time it takes for an REase that is already bound to the DNA to cleave the DNA and release the quantum dot from the glass surface. Quantum dot disappearance happens quickly and results in a sharp decrease in the intensity trajectory, providing a clear indication of the time at which a given DNA molecule is cleaved. The determination of event times is accomplished by mathematical analysis of intensity trajectories, and a typical experiment results in hundreds of identifiable cleavage events. The results of multiple experiments can be pooled to provide adequate statistics to allow estimation of the parameters, N and β, by either nonlinear least-squares or maximum-likelihood analysis.
The flow cell is directly coupled to a high numerical aperture oil-immersion 60x magnification objective on an inverted microscope equipped with laser illumination for through-objective TIRF imaging (Figure 5A). After introducing the DNA substrate and washing away excess DNA and quantum dots, there are typically thousands of individual quantum dots in a field of view (Figure 5B). These quantum dots are stably attached to the glass surface, and they do not undergo noticeable dimming or significant bleaching over the time scale of the experiment. However, if a buffer containing both magnesium and an appropriate REase is flowed through the flow channel, at the end of a typical four-minute observation period, at least 30% of the quantum dots present at the beginning of an experiment will have disappeared from the field of view. Confirming the magnesium requirement, when the REase is flowed through the channel in the absence of magnesium, at least 95% of quantum dots present at the beginning of the experiment can still be seen at the end of the observation period (Figure 5C). However, when magnesium-containing buffer is flowed immediately after allowing the REase to bind the surface-bound DNA in the absence of magnesium, up to half of the quantum dots will have disappeared by the end of the observation period (Figure 5D), similar to what is observed when REase and magnesium were flowed through the channel together. The exact yield of events will depend on the efficiency of the enzyme under the conditions used. When magnesium-containing buffer is flowed without having previously introduced the appropriate REase into the flow channel, less than 5% of quantum dots disappear during the observation period, and there is no discernible peak for a histogram of the events. This result indicates that prebound REase molecules mediate cleavage of the DNA that tethers the quantum dots to the glass surface, and this DNA cleavage is responsible for the vast majority of observed quantum dot disappearance events in these experiments.
Quantum dot disappearance happens quickly and results in a sharp decrease in the intensity trajectory, providing a clear indication of the time at which a given DNA molecule is cleaved (Figure 5E). The determination of event times is accomplished by mathematical analysis of intensity trajectories. Single quantum dots generate intensity trajectories with significantly higher variance than the background under the image conditions used, so putative events are confirmed when the variance of the intensity trajectory decreases to a level comparable to the variance of the background after the observed intensity drop. In addition, trajectories that include a large degree of blinking prior to a putative disappearance event are excluded from the final analysis. However, a typical experiment results in hundreds of events that meet these criteria, and the results of multiple experiments can be pooled to provide adequate statistics to allow estimation of the parameters N and β by either nonlinear least-squares curve-fitting or maximum-likelihood parameter estimation.
The representative data presented were collected by performing this experiment with the well-studied Type IIP REase, EcoRV (Video 1). The 60-bp-long duplex DNA substrate is constructed with a biotin molecule on the 5' end of one strand of the duplex and a quantum dot covalently attached to the 5' end of the other strand. The DNA substrate contains a single copy of the recognition sequence, GAT↓ATC, which is cleaved by EcoRV in the middle, as indicated by the downward arrow (↓). EcoRV was prebound to the DNA substrate in the absence of magnesium, and then magnesium-containing buffer was flowed to initiate DNA cleavage. The representative data include the pooled results of five separate experiments, yielding a total of 3451 observed cleavage events. After excluding events that occurred before the zero time-point or outside of the prominent activity peak, 2987 events remained, which were enough to populate a histogram with one-second bins. Both nonlinear least-squares curve-fitting and maximum-likelihood parameter estimation were used to extract the values of N and β from the data. The two parameter estimation methods were in agreement (Figure 6A), with N = 3.52 (95% confidence interval: 3.32-3.71) and β = 5.78 s (95% confidence interval: 5.41-6.15 s) for the nonlinear least-squares fit and N = 3.41 (95% confidence interval: 3.25-3.58) and β = 6.06 s (95% confidence interval: 5.75-6.39 s) for the maximum-likelihood estimation.
The estimate of at least three kinetic steps is reasonable given what is known about the mechanism of EcoRV. This enzyme is known to bind DNA non-specifically in the absence of magnesium14. Bulk solution phase observations of the changes in the intrinsic fluorescence of tryptophan residues in EcoRV under stopped flow conditions suggested that EcoRV that has bound to DNA in the absence of magnesium must undergo a conformational change prior to the entrance of magnesium into the active site18. This observation is corroborated by crystal structure data19. The results of tryptophan fluorescence studies also indicate that DNA cleavage and product release are separate steps that occur with similar rates18. Therefore, a reasonable reaction mechanism for EcoRV-mediated DNA cleavage in this experiment is:
ES → ES* → EP → E+P
In this mechanism, ES, the initial enzyme-substrate complex, is formed when EcoRV is prebound to the DNA in the absence of magnesium. When magnesium enters the flow cell, the enzyme-substrate complex undergoes a conformational change to become ES*, the activated enzyme-substrate complex. This activated complex then cleaves DNA, but does not immediately release the product molecules, becoming an enzyme-product complex (EP). Finally, the product, P, is released in the final step. This mechanism requires three steps, which is in agreement with the data presented. The resulting estimate for the average waiting time for each step is ~6 s, equivalent to a rate of 0.17 s-1 for each step. This calculation is in general agreement with previous estimates of the rates for these processes-on the order of 0.3-0.4 s-1 for DNA cleavage and product release and ~0.5 s-1 for the conformational rearrangement.
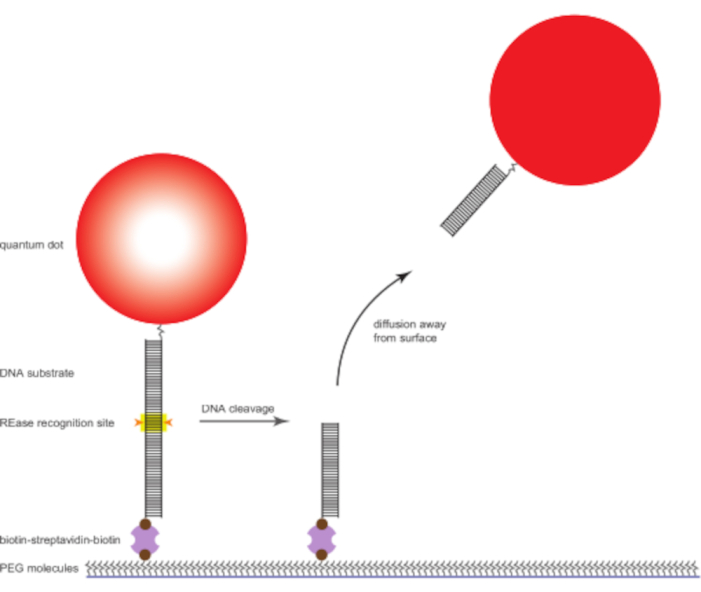
Figure 1: Labeled DNA and enzyme reaction schematic. Quantum-dot-labeled DNA molecules are tethered to the functionalized glass surface via biotin-streptavidin-biotin linkage and observed using TIRF microscopy. The DNA molecules contain the recognition site for the REase of interest. When a DNA molecule is cleaved by the REase, the quantum dot is free to diffuse away from the surface and out of the illumination zone. Abbreviations: REase = restriction endonuclease; PEG = polyethylene glycol; TIRF = total internal reflection fluorescence. Please click here to view a larger version of this figure.
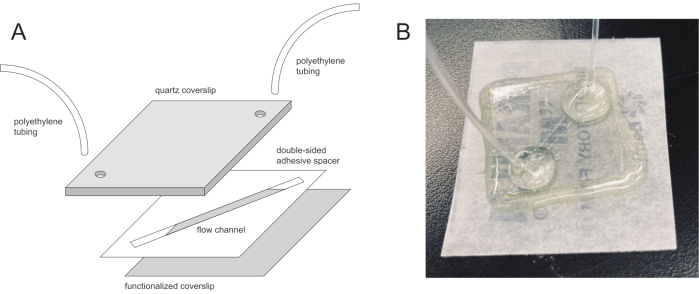
Figure 2: Microfluidic flow cell device. (A) Exploded view showing the three layers used to create the device: the functionalized glass coverslip on the bottom, the quartz slide with inlet and outlet holes on the top, and the double-sided adhesive imaging spacer with a channel cut into it sandwiched in the middle. (B) A completed device with polyethylene tubes sealed in place and with edges coated with epoxy. Please click here to view a larger version of this figure.
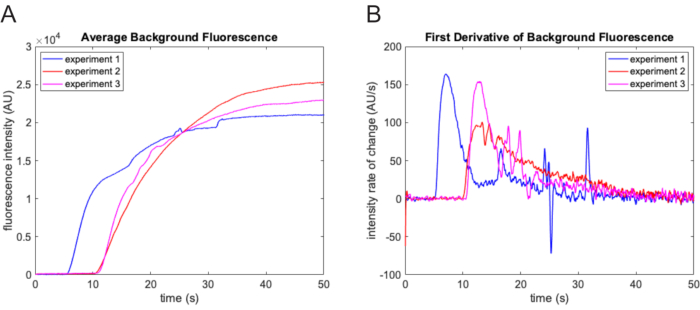
Figure 3: Zero time-point determination. (A) Average background intensity as determined using the morphological top-hat filtering function over each frame of the movie. The background intensity increases markedly when the experimental buffer containing fluorescein enters the field of view. The results from three different experiments are shown here. There is substantial variation in the lag time from experiment to experiment. (B) The sharp increase in the rate of change of the background fluorescence intensity trajectory facilitates accurate determination of the zero time-point. Please click here to view a larger version of this figure.
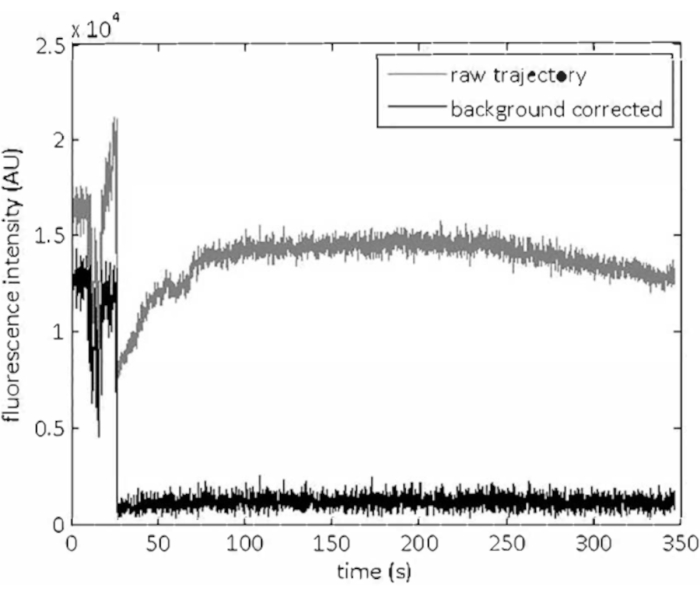
Figure 4: Background correction. The increase in the background intensity due to the fluorescein tracer dye indicating the arrival of magnesium can be seen in the raw fluorescence intensity trajectories for individual quantum dots (grey trajectory). After subtracting background using the morphological top-hat filtering function, the artefacts introduced by the tracer dye have been removed from the trajectory (black trajectory). Please click here to view a larger version of this figure.
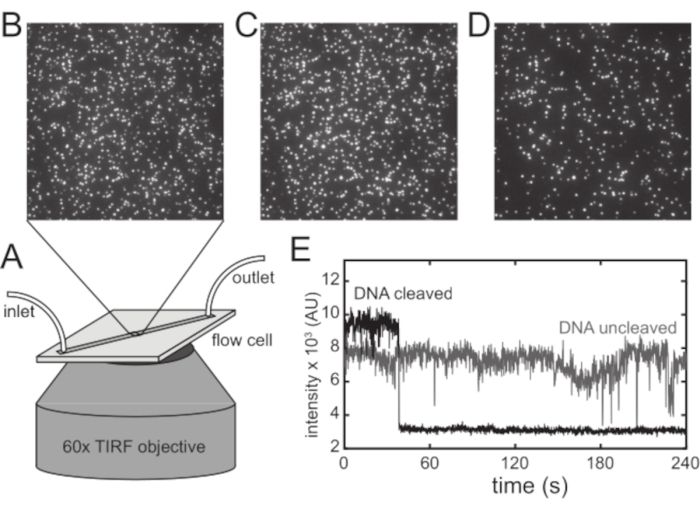
Figure 5: Single-molecule TIRF experiment for parallelized observations of DNA cleavage events. (A) A flow cell constructed on a functionalized glass surface designed to capture quantum-dot-labeled DNA is directly coupled to a high numerical aperture 60x oil immersion microscope objective for TIRF imaging. (B) Representative image from the beginning of an experiment. Quantum-dot-labeled DNA has been loaded into the flow cell, and excess unbound quantum dots have been washed away. Individual DNA-tethered quantum dots can be resolved from one another. (C) The same field of view after buffer containing a REase capable of cleaving the DNA tethers has been flowed through the flow channel in the absence of magnesium for four minutes. There has been no significant loss of DNA-tethered quantum dots. (D) The same field of view at the conclusion of an experiment. Magnesium-containing buffer was flowed through the flow channel immediately after flowing REase in a magnesium-free buffer. This image was acquired after approximately four minutes of buffer flow. Prebound REases have cleaved many of the DNA tethers, releasing the quantum dots from the surface. For ease of viewing, only the central quadrant of the objective field of view is shown in each image. Ten movie frames (equivalent to one second of observation time) were averaged to diminish the effects of quantum dot blinking. Brightness and contrast settings are identical for all three images. (E) Representative fluorescence intensity trajectories from image locations where a quantum dot was present at the beginning of the experiment. Trajectories obtained from image locations corresponding to quantum dots that are not released from the surface (grey) may display brief dips to a lower intensity level, but they begin and end at a high intensity level. Trajectories obtained from image locations, corresponding to quantum dots that are released from the surface (black), display a rapid decrease in intensity to a low background level that is instantaneous with respect to the time resolution of the experiment (10 Hz). Released quantum dots do not reappear within the four-minute observation period. Abbreviations: REase = restriction endonuclease; TIRF = total internal reflection fluorescence. Please click here to view a larger version of this figure.
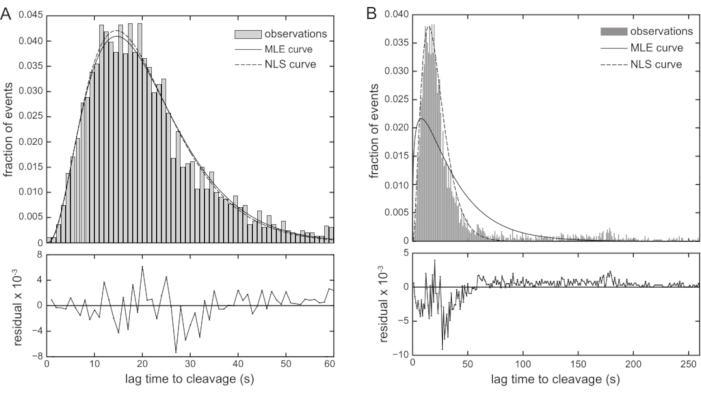
Figure 6: Dwell-time distribution analysis of EcoRV-mediated DNA cleavage. (A) Histogram and predicted envelopes for the 2987 cleavage events that occur within the main activity peak in a set of five pooled experiments with EcoRV. The two predicted curves are nearly identical, and the fit residuals (below the histogram) do not indicate systematic error in the nonlinear least-squares fit. (B) Histogram of the entire set of 3393 cleavage events that occurred after the zero time-point. The curve predicted by the maximum-likelihood estimation of the parameters (unbroken line, MLE curve) assuming a gamma probability distribution fails to enclose the histogram. The curve predicted by a nonlinear least-squares fit of the formula for the gamma probability distribution to the bin heights (dashed line, NLS curve) is superior, but the residuals of the fit (below the histogram) reveal systematic error. Abbreviations: MLE: maximum-likelihood estimation; NLS = nonlinear least-squares. Please click here to view a larger version of this figure.
| Name of Buffer | Component | Concentration | pH at 25 °C |
| sodium phosphate buffer | sodium phosphate | 100 mM | 8.3 |
| CHES buffer | N-cyclohexyl-2-aminoethanesulfonic acid (CHES) | 10 mM | 9.0 |
| PBS | sodium phospate | 3 mM | 7.2 – 7.6 |
| sodium chloride | 150 mM | ||
| potassium phosphate | 1.05 mM | ||
| storage buffer | sodium chloride | 100 mM | 8.0 |
| Tris-HCl | 50 mM | ||
| bovine serum albumin (BSA) | 0.5 mg/mL | ||
| sodium bicarbonate | sodium bicarbonate | 100 mM | 8.2 |
| blocking buffer | Tris-HCl | 20 mM | 7.5 |
| Ethylenediaminetetraacetic acid (EDTA) | 2 mM | ||
| sodium chloride | 50 mM | ||
| Tween-20 | 0.005% (v/v) | ||
| bovine serum albumin (BSA) | 0.2 mg/mL | ||
| experimental buffer without magnesium | sodium chloride | 100 mM | 7.9 |
| Tris-HCl | 50 mM | ||
| dithiothreitol (DTT) | 1 mM | ||
| experimental buffer with magnesium | sodium chloride | 100 mM | 7.9 |
| Tris-HCl | 50 mM | ||
| magnesium chloride | 10 mM | ||
| dithiothreitol (DTT) | 1 mM | ||
| fluorescein | adjust according to imaging conditions | ||
| DNase buffer | Magnesium chloride | 2.5 mM | 7.6 |
| Tris-HCl | 10 mM | ||
| calcium chloride | 0.1 mM |
Table 1: Table of Buffers.
Video 1: Example single-molecule movie. Please click here to download this video.
