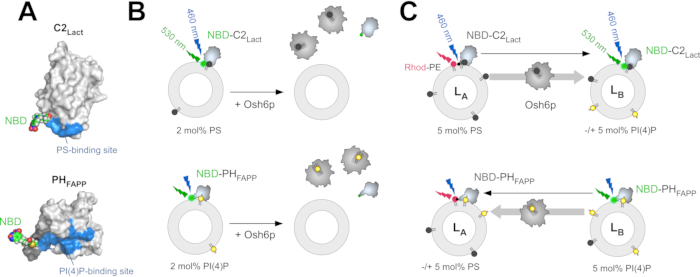
Figure 1: Description of the fluorescent lipid sensors and in vitro assays. (A) Three-dimensional models of NBD-C2Lact and NBD-PHFAPP based on the crystal structure of the C2 domain of bovine lactadherin (PDB ID: 3BN648) and the NMR structure of the PH domain of the human FAPP1 protein (PDB ID: 2KCJ46). An N,N'-dimethyl-N-(thioacetyl)-N'-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)ethylenediamine moiety, built manually and energetically minimized, was grafted onto the thiol function of C352 (NBD-C2Lact) and C13 (NBD-PHFAPP) residues (represented as spheres, with carbon in green, nitrogen in blue, oxygen in red, sulfur in yellow, and hydrogen in white). The surface of the lipid-binding site of each probe was colored in blue. (B) Extraction assays. In the PS extraction assay, PC/PS liposomes (98/2 mol/mol) were incubated with 250 nM NBD-C2Lact. In the absence of extraction, the probe strongly bound to the liposomes, resulting in a blue shift of NBD fluorescence and an increase of its emission intensity. If PS extraction occurred in the presence of an LTP (e.g., Osh6p), the probe dissociated from the liposomes, and its fluorescence was lower. In the PI(4)P extraction assay, liposomes were doped with 2 mol% PI(4)P, and 250 nM NBD-PHFAPP was used. (C) FRET-based lipid transport assays. In the PS transport assay, PC/PS/Rhod-PE liposomes (93/5/2 mol/mol, LA) were incubated with 250 nM NBD-C2Lact. PC liposomes (LB), doped or not with 5 mol% PI(4)P, and Osh6p were sequentially added at t = 1 min and t = 4 min, respectively. If PS transport occurs, this elicits a dequenching of the NBD signal corresponding to the translocation of NBD-C2Lact from LA to LB liposomes. In the PI(4)P transport assay, LB liposomes doped with 5 mol% PI(4)P were incubated with 250 nM NBD-PHFAPP. PC liposomes (LA) doped or not with 5 mol% PS were added. If PI(4)P transport occurs, this causes a quenching of the NBD signal due to the translocation of NBD-PHFAPP from LB to LA liposomes. Abbreviations: NBD = 7-nitrobenz-2-oxa-1,3-diazol fluorophore; NMR = nuclear magnetic resonance; FAPP1 = four-phosphate-adaptor protein 1; PDB = Protein Data Bank; PS= phosphatidylserine; PC = phosphatidylcholine; LTP = lipid transfer protein; Osh6p = oxysterol-binding protein (OSBP) homolog 6 protein; PI(4)P = phosphatidylinositol 4-phosphate; FRET = fluorescence resonance energy transfer; Rhod-PE = rhodamine-labelled phosphatidylethanolamine; LA liposomes = liposomes composed of PC, doped with 2 mol% Rhod-PE, and containing or not 5 mol% PS; LB liposomes = liposomes incorporating 5 mol% PI(4)P. Please click here to view a larger version of this figure.
Figure 2A shows an SDS-PAGE analysis of the products of different steps leading to the purification of C2Lact. Lane 1 shows the protein profile of the lysed bacteria expressing GST-C2Lact (~44.8 kDa), whereas lanes 2 and 3 respectively show the protein profiles of the supernatant and bacterial debris after ultracentrifugation. The comparison of these lanes shows that GST-C2Lact has been recovered in the supernatant and thus can be isolated using glutathione-linked agarose beads. Lanes 4 and 5 show the protein profiles of the supernatant after incubation with beads and washes recovered by gravity flow, whereas lane 6 shows the profile of proteins that have been retained on the beads. An analysis of these lanes indicates that almost all GST-C2Lact has been recovered from the beads.
Lanes 8-12 show the presence of a major band corresponding to C2Lact (~17.9 kDa) in the supernatants recovered through successive washes of the beads after thrombin treatment. Lane 13 indicates that non-cleaved GST-C2Lact, along with GST (~26.9 kDa), remained bound to beads after this treatment. The comparison of these lanes indicates that the cleavage procedure, albeit not 100% efficient, did yield C2Lact that was then fluorescently labelled. Figure 2B shows an ultraviolet (UV)-visible absorbance spectrum of C2Lact labelled with NBD. As the construct is 100% pure, these results confirm that all C2Lact molecules were labelled with an NBD group based on the optical density measured at 280 nm (Trp) and 495 nm (NBD). The purity of NBD-C2Lact and its fluorescence were determined by SDS-PAGE analysis (Figure 2C).
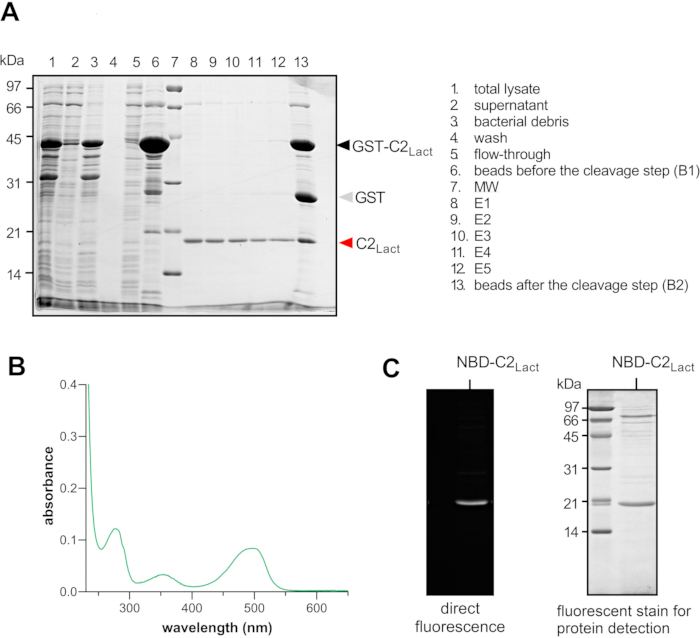
Figure 2: NBD-C2Lact purification. (A) SDS-PAGE analysis was used to check the presence of the protein at different steps of the purification procedure before labeling. The arrowheads indicate the position of the C2Lact domain (red arrowhead), of the GST alone (grey arrowhead), and of the GST-C2Lact construct (black arrowhead). (B) UV-visible absorbance spectrum of NBD-C2Lact. (C) SDS-PAGE analysis of the purified NBD-C2Lact. The first image was acquired under UV illumination without staining and reveals the presence of the NBD-C2Lact construct as it emits fluorescence. The second image shows the same gel after a protein staining procedure (see the Table of Materials). Abbreviations: NBD = 7-nitrobenz-2-oxa-1,3-diazol fluorophore; NBD-C2Lact = N,N'-dimethyl-N-(thioacetyl)-N'-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)ethylenediamine moiety linked to the thiol function of C352 residue of a reengineered version of the C2 domain of bovine lactadherin (PDB ID: 3BN648); PDB = Protein Data Bank; SDS-PAGE = sodium dodecylsulfate polyacrylamide gel electrophoresis; GST = glutathione S-transferase; MW = molecular-weight size marker; UV = ultraviolet. Please click here to view a larger version of this figure.
Figure 3 shows the results from PS and PI(4)P extraction assays using Osh6p. When only incubated with liposomes containing 2 mol% PS, the fluorescence of NBD-C2Lact was maximal as the NBD fluorophore was inserted in the membrane (i.e., a hydrophobic context), indicating that the sensor was membrane-bound. In the presence of Osh6p, the fluorescence was lower and comparable to that measured with pure PC liposomes (Figure 3A). The normalization of intensity values at 536 nm indicated that ~75% of accessible PS was extracted. In the second assay, NBD-PHFAPP was mixed with liposomes containing 2 mol% PI(4)P. The NBD signal was high without Osh6p, but low when Osh6p was present and was similar to that measured with PI(4)P-free liposomes (Figure 3B). An analysis of the intensity revealed that ~100 % of accessible PI(4)P was extracted by the LTP.
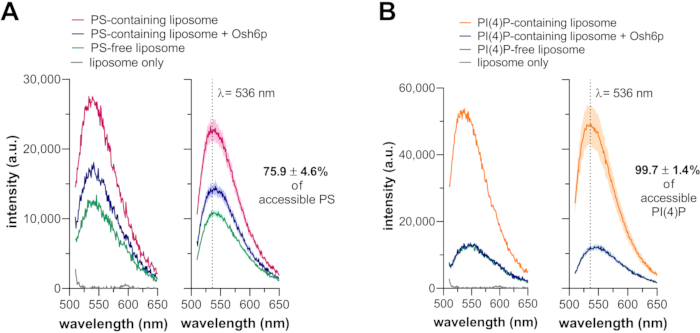
Figure 3: Extraction assays. (A) Fluorescence spectra of NBD-C2Lact (250 nM) measured upon excitation at 460 nm in the presence of liposomes (80 µM, 2 mol% PS) in the absence or presence of 3 µM Osh6p. Reference spectra were recorded with pure PC liposomes incubated or not with NBD-C2Lact (left panel). Several spectra were recorded from different series of wells, corrected by subtracting the background scattering signal from DOPC liposomes alone and averaged (n=4, ± SEM). The percentage of accessible PS that was extracted is indicated (right panel). (B) Fluorescence spectra of NBD-PHFAPP (250 nM) mixed with liposomes (80 µM, 2 mol% PI(4)P) in the absence or presence of 3 µM Osh6p. Reference spectra recorded with PC liposomes in the presence and absence of the sensor are shown (left panel). Several spectra were recorded from different series of wells, corrected by subtracting the background scattering signal from DOPC liposomes alone and averaged (n=4, ± SEM). The percentage of accessible PI(4)P that was extracted is indicated (right panel). Abbreviations: NBD = 7-nitrobenz-2-oxa-1,3-diazol fluorophore; NBD-C2Lact = N,N'-dimethyl-N-(thioacetyl)-N'-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)ethylenediamine moiety linked to the thiol function of C352 residue of a reengineered version of the C2 domain of bovine lactadherin (PDB ID: 3BN648); PDB = Protein Data Bank; PS= phosphatidylserine; PC = phosphatidylcholine; DOPC = dioleoylphosphatidylcholine; Osh6p = oxysterol-binding protein (OSBP) homolog 6 protein; PI(4)P = phosphatidylinositol 4-phosphate; SEM = standard error of the mean; a.u. = arbitrary units. Please click here to view a larger version of this figure.
Figure 4A shows typical results from a PS transfer assay using Osh6p as an LTP. At time zero, NBD-C2Lact was mixed with LA liposomes containing 5 mol% 16:0/18:1-PS (POPS) and 2 mol% Rhod-PE in a volume of 570 µL of HKM buffer at 30 °C. As the probe bound to LA liposomes, its signal was quenched due to FRET with Rhod-PE present in these liposomes. After one minute, Rhod-PE-free LB liposomes (30 µL) were added; this was expected to only elicit a slight change in the signal due to light diffusion by this second liposome population and/or a dilution effect. The signal intensity after the addition of the LB liposomes corresponds to F0. The injection of a few µL of a stock solution of Osh6p (typically 40 µM) to dilute 200 nM of the protein in the reaction mix elicited a slow increase in the NBD signal due to the dequenching of the fluorophore as PS was transported from LA to LB liposomes, thereby promoting the translocation of NBD-C2Lact.
When LB liposomes contained 5 mol% PI(4)P, the dequenching was much faster, as PS was transferred more rapidly by Osh6p to LB liposomes due to the counterexchange of PS with PI(4)P by the LTP (second curve). The third curve corresponds to an experiment in which NBD-C2Lact was mixed with equal amounts of LA-Eq and LB-Eq liposomes. The signal was higher than F0 and corresponded to a situation where the probe was evenly bound to LA and LB liposomes, thus reflecting a situation where PS was fully equilibrated between the two populations of liposomes. FEq was calculated by averaging the value of the signal measured in the last 5 min of the experiment.
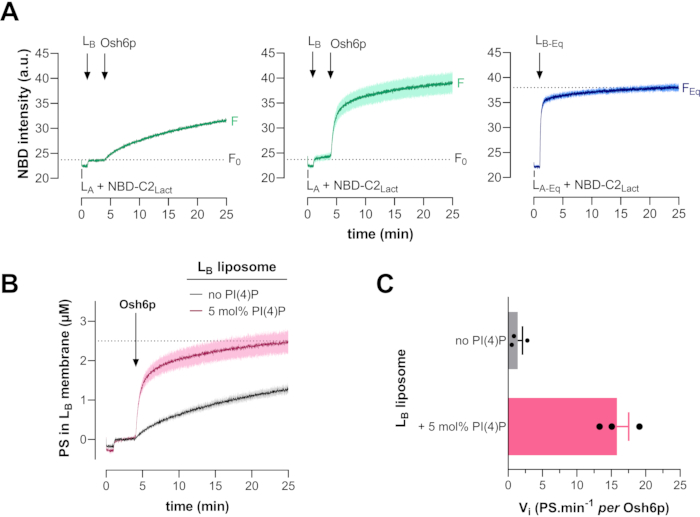
Figure 4: Typical PS transport kinetics measured with Osh6p and reference curve. (A) LB liposomes devoid of PI(4)P (200 µM total lipids) and Osh6p (200 nM) were sequentially added to a cuvette containing NBD-C2Lact (250 nM) and LA liposomes (200 µM) doped with 5 mol% PS and 2 mol% Rhod-PE (left curve). The same experiment was performed with LB liposomes doped with 5 mol% PI(4)P (middle curve). To determine FEq, NBD-C2Lact (250 nM) was premixed with LA-Eq liposomes; then, LB-Eq liposomes were added (right curve). (B) Averaged PS transport curves determined after the normalization of several measurements done using LB liposomes with or without PI(4)P (mean ± SEM, n=3). (C) Initial PS transfer rates (mean ± SEM, n=3). Abbreviations: NBD = 7-nitrobenz-2-oxa-1,3-diazol fluorophore; NBD-C2Lact = N,N'-dimethyl-N-(thioacetyl)-N'-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)ethylenediamine moiety linked to the thiol function of C352 residue of a reengineered version of the C2 domain of bovine lactadherin (PDB ID: 3BN648); PDB = Protein Data Bank; PS= phosphatidylserine; Osh6p = oxysterol-binding protein (OSBP) homolog 6 protein; PI(4)P = phosphatidylinositol 4-phosphate; Rhod-PE = rhodamine-labelled phosphatidylethanolamine; LA liposomes = liposomes composed of phosphatidylcholine, doped with 2 mol% Rhod-PE, and containing or not 5 mol% PS; LB liposomes = liposomes incorporating 5 mol% PI(4)P; F = fluorescence; F0 = fluorescence corresponding to NBD before addition of Osh6p; FEq = fluorescence signal if PS is fully equilibrated between LA and LB liposomes by a transfer process; SEM = standard error of the mean. Please click here to view a larger version of this figure.
Figure 4B shows average kinetic curves of PS transfer from LA liposomes to LB liposomes, doped or not doped with PI(4)P after the normalization of F data using F0 and FEq as reference values. The initial transport rate for each experiment was calculated by fitting the initial data points measured after the injection of the protein with a linear function. Figure 4C shows the mean initial PS transport rate determined from three distinct experiments using LB liposomes with or without PI(4)P. When LB liposomes contained 0 and 5 mol% PI(4)P, the rates were respectively equal to 1.4 and 15.8 PS.min-1 per Osh6p molecule. Figure 5A shows typical results from a PI(4)P transfer assay using Osh6p as an LTP, which was carried out with the same materials and conditions as those used for the PS transport assay.
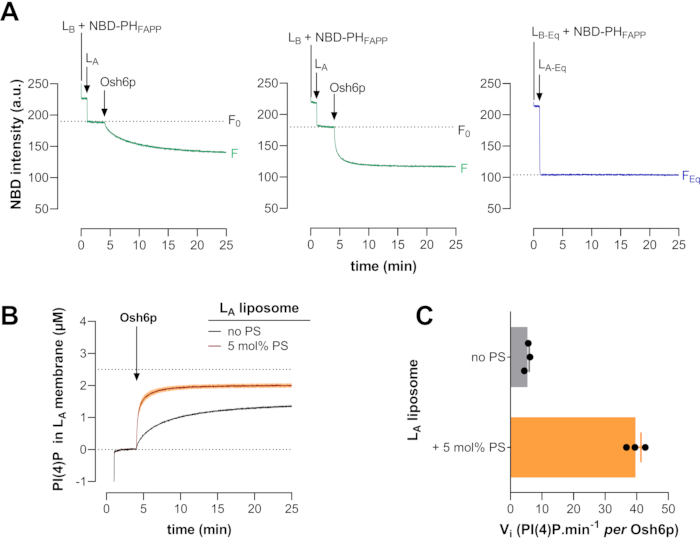
Figure 5: Typical PI(4)P transport kinetics measured with Osh6p and reference curve. (A) PS-free liposomes containing 2 mol% Rhod-PE (LA, 200 µM total lipids) and Osh6p (200 nM) were sequentially added to a cuvette containing NBD-PHFAPP (250 nM) and LB liposomes (200 µM) doped with 5 mol% PI(4)P (left curve). The same experiment was performed with LA liposomes doped with 5 mol% PS (middle curve). To determine FEq, NBD-PHFAPP (250 nM) was premixed with LB-Eq liposomes; then, LA-Eq liposomes were added (right curve). (B) PI(4)P transfer kinetics determined after normalization of several measurements of LA liposomes with or without PS (mean ± SEM, n=3). (C) Initial PI(4)P transfer rates (mean ± SEM, n=3). Abbreviations: NBD = 7-nitrobenz-2-oxa-1,3-diazol fluorophore; NBD-PHFAPP = NBD-labeled Pleckstrin homology domain of the human four-phosphate-adaptor protein 1 (FAPP1, UniProt: Q9HB20, segment [1-100]); PS= phosphatidylserine; Osh6p = oxysterol-binding protein (OSBP) homolog 6 protein; PI(4)P = phosphatidylinositol 4-phosphate; Rhod-PE = rhodamine-labelled phosphatidylethanolamine; LA liposomes = liposomes composed of phosphatidylcholine, doped with 2 mol% Rhod-PE, and containing or not 5 mol% PS; LB liposomes = liposomes incorporating 5 mol% PI(4)P; F = fluorescence; F0 = fluorescence corresponding to NBD before addition of Osh6p; FEq = fluorescence signal if PI(4)P is fully equilibrated between LA and LB liposomes by a transfer process; SEM = standard error of the mean. Please click here to view a larger version of this figure.
At time zero, NBD-PHFAPP was mixed with LB liposomes containing 5 mol% PI(4)P in a volume of 570 µL of HKM buffer. Because NBD-PHFAPP was bound to LB liposomes, its signal was high. After one minute, LA liposomes (30 µL) were added, which was expected to only elicit a slight change in the signal. The intensity then corresponded to F0. Injecting Osh6p (200 nM final concentration) into the reaction mix triggered a quenching of the NBD signal due to the translocation of NBD-PHFAPP molecules to LA liposomes as PI(4)P was transferred from LB liposomes to LA liposomes. When LA liposomes contained 5 mol% POPS, the dequenching was much faster owing to a faster PI(4)P transfer resulting from PS/PI(4)P exchange. The third curve corresponds to an experiment in which NBD-PHFAPP was mixed with equal amounts of LA-Eq and LB-Eq liposomes.
The signal was lower than F0 as it corresponded to a situation where the probe was uniformly bound to LA and LB liposomes, thus indicative of a full equilibration of PI(4)P between the liposomes. FEq was calculated by averaging the value of the signal measured in the last 5 min of the experiment. Figure 5B,C show averaged kinetics curves obtained after signal normalization and mean PI(4)P initial transfer rates measured with LA liposomes that were doped or not doped with 5 mol% PS. Here, it is worth noting that both lipid ligands were transported faster and with similar velocities when each lipid was initially present in LA and LB membranes, respectively, which indicates that Osh6p is a PS/PI(4)P exchanger.
