We describe here a protocol to genetically engineer T cells, that can be used to generate both autologous and allogeneic CAR-T cells, as well as TCR redirected T cells.
Figure 1 provides a detailed description of the stages involved in the process of manufacturing CRISPR edited T cells. The process begins by designing sgRNA to the gene of interest. Once the sgRNA are designed and synthesized they are then used to make RNP complexes with the appropriate Cas protein. T cells are isolated from either a healthy donor or a patient apheresis and RNP complexes are delivered either by electroporation or nucleofection. Post editing, the T cells are activated and transduced with the lentiviral vector coding for the CAR or the TCR construct. After activation, T cells are expanded in culture and cryopreserved for future studies. The detailed protocol followed in the laboratory is described in Figure 2. During the expansion, the population doubling and volume changes are tracked throughout the protocol and an example is shown in Figure 2B and C for both mock and edited CAR-T cells. Figure 2B,C show that the KO of the gene of interest did not cause any significant changes in the proliferation and activation during the expansion. These results, however, depend on the target gene being edited and hence may or may not lead to changes in proliferation and expansion.
Once the cells are cryopreserved, levels of CAR expression are also determined for further functional studies. In this case, as shown in Figure 2D we checked CD19 CAR expression on both the Mock edited and KO CAR-T cells and did not see any significant changes. This will again depend on the gene of interest being edited. Finally, the KO efficiency can be determined using multiple techniques such as flow cytometry and western blot for protein level detection and also Sanger sequencing for gene level detection of the KO. Figure 3A and 3B show representative flow cytometry plots where PDCD1 and TRAC locus is targeted using sgRNA, showing an efficiency of 90% for the PDCD1 sgRNA and 98% for the TRAC sgRNA across multiple healthy donors. Thus, this protocol can achieve high efficiency knockout with minimal loss in viability.
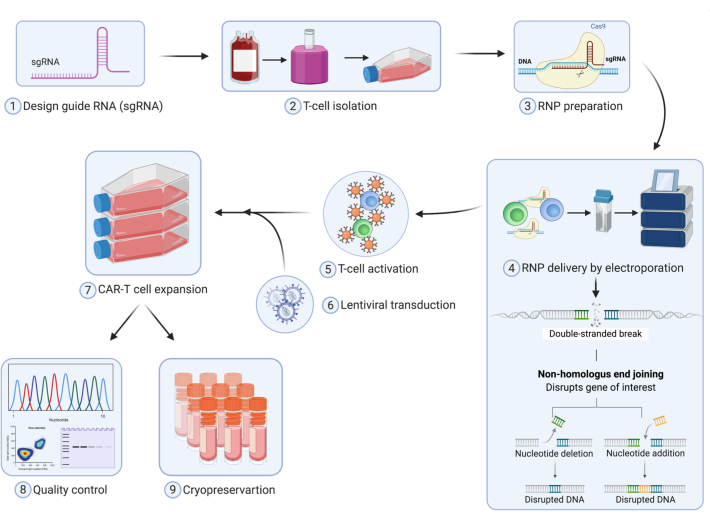
Figure 1. Schematic diagram showing T cell editing using CRISPR Cas9 Technology and manufacturing of primary human CAR-T cells. Please click here to view a larger version of this figure.
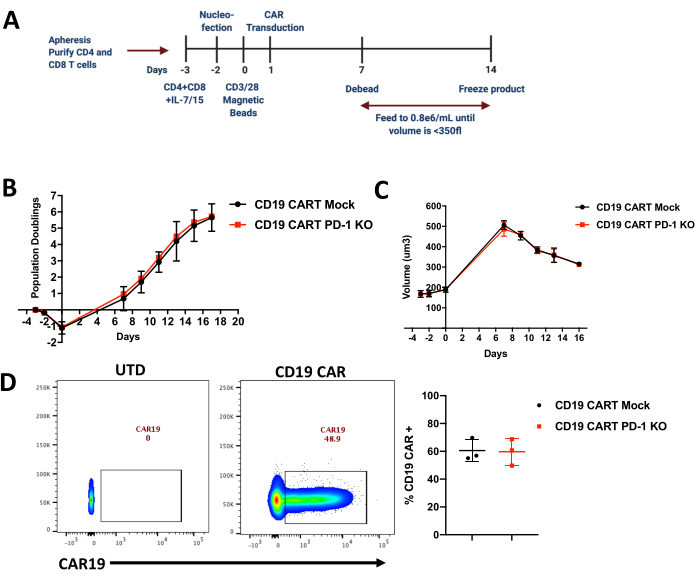
Figure 2. Expansion of edited CAR-T cells and their population doublings. (A) Timeline of CRISPR editing and manufacturing in primary human CART cells. (B) Population Doublings in Mock and CRISPR edited CD19 CAR-T cells measured using a Coulter Counter during the expansion of the CAR-T cells (n=3 healthy donors; KO=knockout) (C) Cell size (µm3) measured using a Coulter Counter during the expansion of the CAR-T cells (n=3 healthy donors). (D) Representative flow cytometry plots showing CAR staining and average in multiple donors showing CAR expression in both mock and edited CAR-T cells. CAR expression was detected using an anti-idiotype antibody conjugated to a fluorophore and gated on Lymphocytes/Singlets/Live Cells (n=3 healthy donors, UTD=untransduced,). Please click here to view a larger version of this figure.
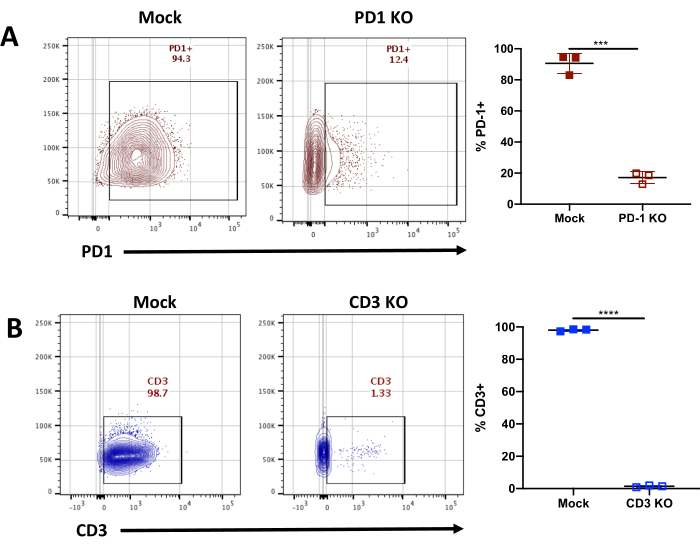
Figure 3. Characterizing KO efficiency in edited CAR-T Cells using flow cytometry. (A) Representative flow cytometry plots showing PD-1 staining and average in multiple donors showing PD-1 KO efficiency using a gRNA targeting the TRAC locus in mock and edited CAR-T cells. PD-1 expression was detected using PD-1 antibody (Clone EH12.2H7) conjugated to fluorophore and gated on Lymphocytes/Singlets/Live Cells (n=3 healthy donors). Error bars indicate mean±standard error of the mean (SEM). **** p<0.0001, *** p=0.0005, ** p=0.001 by Welch's t test. (B) Representative flow cytometry plots showing CD3 staining and average in multiple donors showing CD3 KO efficiency using a gRNA targeting the TRAC locus in mock and edited CAR-T cells. CD3 expression was detected using CD3 antibody (Clone OKT3) conjugated to fluorophore and gated on Lymphocytes/Singlets/Live Cells (n=3 healthy donors). Error bars indicate mean±standard error of the mean (SEM). **** p<0.0001, *** p=0.0005, ** p=0.001 by Welch's t test. Please click here to view a larger version of this figure.
