After the isolation of sEVs from RAW 264.7 conditioned media via centrifugation, NTA was used to determine the concentration and size distribution of the purified sEVs. The average mean size of RAW 264.7-derived sEVs was 140 nm, and the peak particle size was 121.8 nm, confirming that most detectable particles in the light scattering measurement fell within the size range of exosomes or sEVs at 50-150 nm (Figure 1A). As suggested in the minimal information for studies of extracellular vesicles 2018 (MISEV2018)23, we analyzed a set of proteins that should be present or excluded from distinct EV populations. Western blotting of sEVs, cell lysate, and exo-depleted media demonstrated that sEV-derived protein samples contained the sEV marker proteins Alix, CD81, and GAPDH. The cell lysate fraction was enriched with the endoplasmic reticulum resident protein, calnexin, which was absent in the sEVs. Thus, calnexin served as a negative marker for cellular contamination (Figure 1B).
We next performed dose-response and time-course experiments for sEV uptake in vitro. Neuro-2a cells were incubated with a single 1 µg dose of PKH67-labeled sEVs for 1, 4, and 24 h, following which the uptake of different concentrations of sEVs (1, 5, and 10 µg) was examined at 1 h. The results of the NTA indicated that 1 µg of protein on average was equal to ~1 x 109 particles. In parallel, PBS, unlabeled sEVs, and dye-alone controls were also tested. We observed that uptake of sEVs occurred at 1 h (Figure 2A) and for the 1, 5, and 10 µg sEVs (Figure 2B). Fluorescence could be detected at 4 h for 5 and 10 µg of sEVs (Figure 2C) post incubation. Next, uptake of PKH26-labeled sEVs by primary astrocytes was examined (Figure 3). Maximal fluorescence from sEV uptake in primary cortical astrocytes occurred at 24 h. Unlabeled sEVs did not show fluorescence, demonstrating that sEV autofluorescence does not significantly contribute to false positives (Supplemental Figure S1A).
Next, labeled sEVs were intrathecally injected into mice to assess the delivery and uptake of sEVs by different cells in the spinal cord using immunohistochemistry and confocal microscopy. We stained for MAP2 as a neuronal marker, GFAP as an astrocytic marker, and IBA1 as a microglial marker. Neurons (Figure 4), astrocytes (Figure 5), and microglial cells (Figure 6) all took up PKH26-labeled sEVs, and maximal sEV fluorescence was observed at 6 h post-injection. While the sEVs did not always colocalize with the cellular markers, we did not observe any differential uptake by CNS cells. Intrathecal injection with 5 µg of unlabeled RAW 264.7 sEVs or dye control did not show significant fluorescence (Supplemental Figure S1B). Fluorescent signals were observed in the meninges, both 6 h and 18 h after the injection of sEVs (Supplemental Figure S1C).
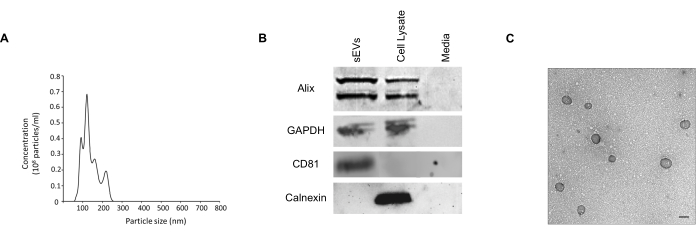
Figure 1: Characterization of purified RAW 264.7 sEVs. (A) Size and concentration of sEVs were determined using NanoSight NS300. The particles were tracked and sized based on Brownian motion and diffusion coefficient. The size distribution of sEVs is shown in nm. The concentration of sEVs was expressed as particles/mL. (B) Western blot of proteins derived from purified sEVs, cell lysate, and exosome-depleted media using sEV markers ALIX, GAPDH, and CD81. The endoplasmic reticulum protein marker, calnexin, serves as a control to monitor cellular contamination in sEV preparations. (C) Transmission electron microscopy demonstrated the size and morphology of sEVs. Scale bar = 100 nm. Abbreviations: sEVs = small extracellular vesicles; ALIX = Alpha-1,3/1,6-Mannosyltransferase (ALG-2)-interacting protein X; GAPDH = glyceraldehyde 3-phosphate dehydrogenase; CD81 = cluster of differentiation 81. Please click here to view a larger version of this figure.
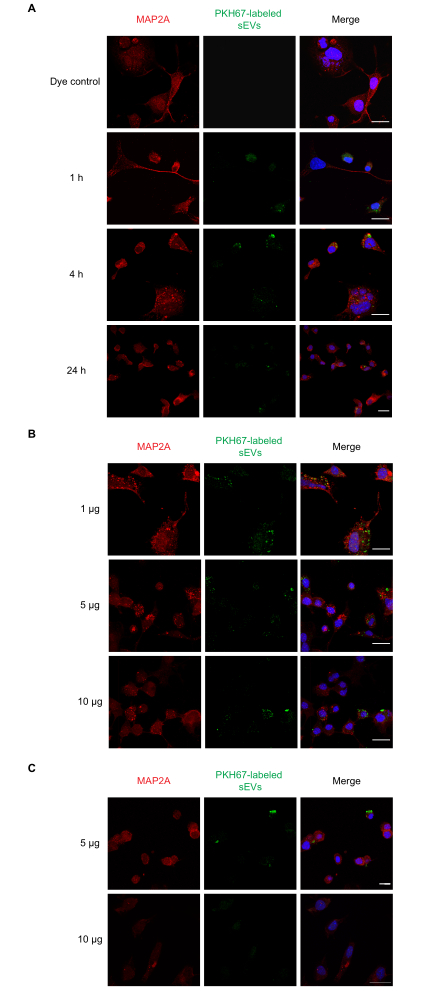
Figure 2: Uptake of labeled RAW 264.7 sEVs by Neuro-2a cells. (A) PKH67-labeled sEVs (1 µg) were added to the cultured Neuro-2a cells for 1, 4, or 24 h. sEV uptake was observed at all time points with confocal microscopy. (B) PKH67-labeled sEVs (1, 5, or 10 µg) were added to Neuro-2a cells for 1 h. (C) PKH67-labeled sEVs (5 or 10 µg) were added to Neuro-2a cells for 4 h. sEV uptake was observed in all dosage groups with confocal microscopy. Negative control groups treated with PKH dye alone did not show sEV staining (Supplemental Figure S1). Neuro-2a cells were immunostained with MAP2A (probed with Alexa Fluor 594, shown in red), while cell nuclei were stained with DAPI (shown in blue) and sEVs with PKH67 (shown in green). Scale bar = 50 µm. Abbreviations: sEVs = small extracellular vesicles; MAP2A = microtubule-associated protein 2A; DAPI = 4′,6-diamidino-2-phenylindole. Please click here to view a larger version of this figure.
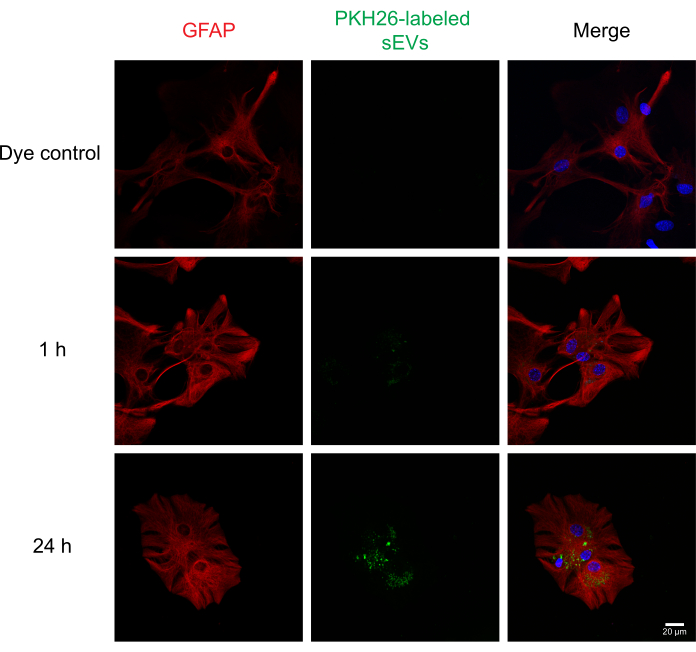
Figure 3: Uptake of PKH26-labeled RAW 264.7 sEVs by primary mouse cortical astrocytes. One µg of sEVs was labeled with PKH26 dye and added to the primary astrocyte culture medium. Uptake of sEVs was observed at 1 and 24 h post-addition using a confocal laser scanning microscope. Astrocytes were stained with GFAP (probed with Alexa Fluor 488, shown in red), while cell nuclei were counterstained with DAPI (shown in blue), and sEVs were previously stained with PKH26 (shown in green). Scale bar = 20 µm. PKH26 dye alone served as a negative control for sEV staining. Abbreviations: sEVs = small extracellular vesicles; GFAP = glial fibrillary acidic protein; DAPI = 4′,6-diamidino-2-phenylindole. Please click here to view a larger version of this figure.
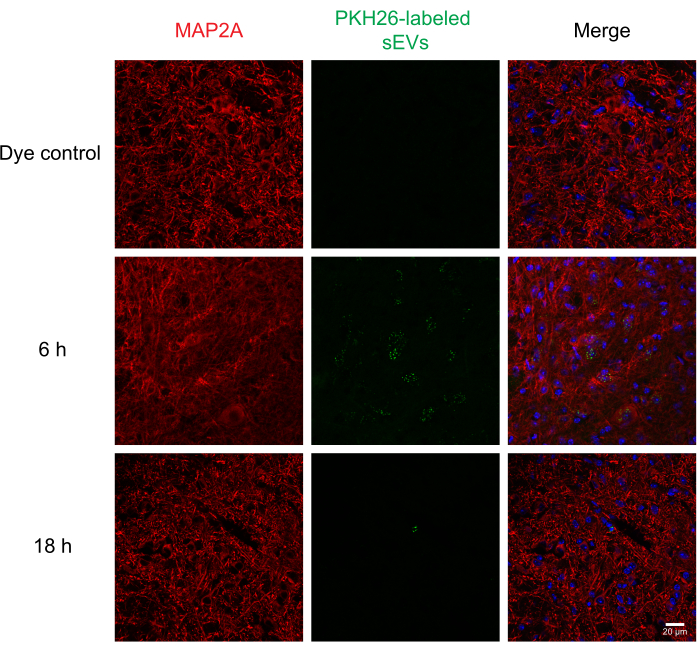
Figure 4: Uptake of RAW 264.7 sEVsin neurons. PKH26-labeled sEVs were injected intrathecally in mice; 6 and 18 h later, the mice were perfused with 4% PFA, and the spinal cord was isolated and sectioned at 30 µm. Spinal cord sections were immunostained with a cell marker (probed with Alexa Fluor 488, shown in red) and DAPI nuclear counterstain (shown in blue), while sEVs were previously labeled with PKH26 (shown in green). Spinal cord sections were immunostained for MAP2A to visualize the neurons (red). Confocal microscopy shows sEVs in MAP2A-positive neurons at different time points. The negative control, PKH26 dye-alone group, did not show sEV staining. Scale bar = 20 µm. Abbreviations: sEVs = small extracellular vesicles; PFA = paraformaldehyde; MAP2A = microtubule-associated protein 2A; DAPI = 4′,6-diamidino-2-phenylindole. Please click here to view a larger version of this figure.
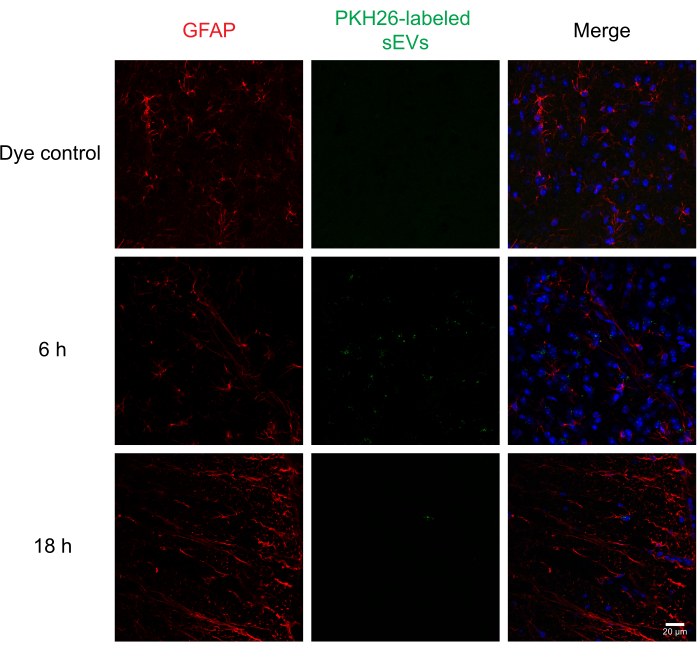
Figure 5: Uptake of RAW 264.7 sEVs in astrocytes. PKH26-labeled sEVs were injected intrathecally in mice; 6 and 18 h later, the mice were perfused with 4% PFA, and the spinal cord was isolated and sectioned at 30 µm. Spinal cord sections were immunostained with a cell marker (probed with Alexa Fluor 488, shown in red) and DAPI nuclear counterstain (shown in blue), while sEVs were previously labeled with PKH26 (shown in green). Spinal cord sections were immunostained for GFAP to visualize the astrocytes (red). Confocal microscopy shows sEVs in GFAP-positive astrocytes at different time points. The negative control, PKH26 dye-alone group, did not show sEV staining. Scale bar = 20 µm. Abbreviations: sEVs = small extracellular vesicles; PFA = paraformaldehyde; GFAP = glial fibrillary acidic protein; DAPI = 4′,6-diamidino-2-phenylindole. Please click here to view a larger version of this figure.
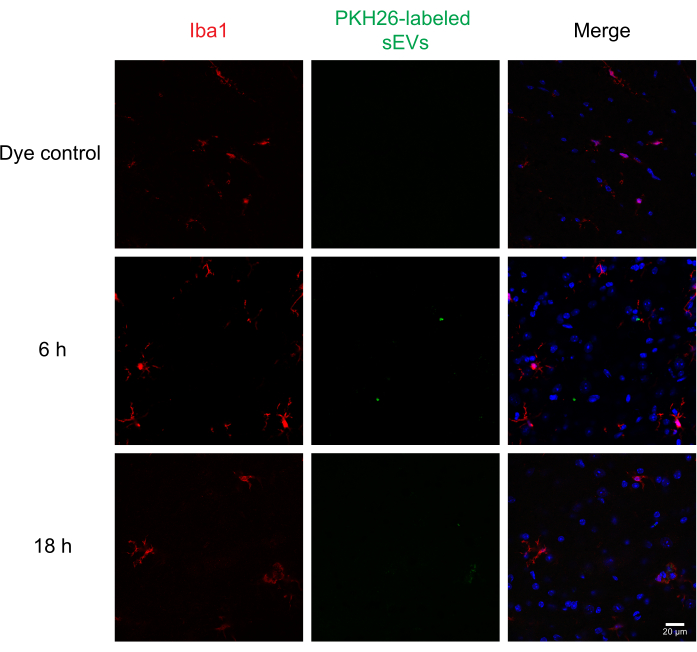
Figure 6: Uptake of RAW 264.7 sEVs in microglia. PKH26-labeled sEVs were injected intrathecally in mice; 6 and 18 h later, the mice were perfused with 4% PFA, and the spinal cord was isolated and sectioned at 30 µm. Spinal cord sections were immunostained with a cell marker (probed with Alexa Fluor 488, shown in red) and DAPI nuclear counterstain (shown in blue), while sEVs were previously labeled with PKH26 (shown in green). Spinal cord sections were immunostained for IBA1 to visualize the microglia (red). Confocal microscopy shows sEVs in IBA1-positive microglia at different time points. The negative control, PKH26 dye-alone group, did not show sEV staining. Scale bar = 20 µm. Abbreviations: sEVs = small extracellular vesicles; PFA = paraformaldehyde; IBA1 = ionized calcium-binding adaptor molecule 1; DAPI = 4′,6-diamidino-2-phenylindole. Please click here to view a larger version of this figure.
Supplemental Figure S1: Uptake of labeled RAW 264.7 sEVs by primary mouse cortical astrocytes and in spinal cord. (A) Controls for the uptake of PKH26-labeled RAW 264.7 sEVs by primary mouse cortical astrocytes. One µg of unlabeled sEVs resuspended in PBS or an equal volume of PBS was added in parallel to the culture medium of astrocytes. No fluorescence was observed at 1 h for PBS and the unlabeled control using a confocal laser scanning microscope. Astrocytes were stained with GFAP (probed with Alexa Fluor 488, shown in red), while the nuclei were counterstained with DAPI (blue), and unlabeled sEVs were visualized under the same Alexa Fluor 546 channel as PKH26-labeled sEVs. Scale bar = 50 µm. (B) Controls for the uptake of PKH26-labeled RAW 264.7 sEVs by mouse spinal cord in vivo. Five µg of unlabeled sEVs or dye control were injected intrathecally in mice. Again, fluorescent signals were not observed for unlabeled sEVs or dye-alone control using a confocal laser scanning microscope. Astrocytes were stained with GFAP (probed with Alexa Fluor 488, shown in red), while nuclei were counterstained with DAPI (blue), and unlabeled sEVs were visualized under the same Alexa Fluor 546 channel as PKH26-labeled sEVs. Scale bar = 50 µm. (C)Representative images reveal the presence of RAW 264.7 sEVs in mouse spinal meninges 6 h and 18 h after intrathecal delivery. Five µg of sEVs were labeled with PKH26 dye (shown in green), and the nuclei were counterstained with DAPI (shown in blue). Asterisks indicate the anterior spinal artery. Scale bar = 50 µm. Abbreviations: sEVs = small extracellular vesicles; PBS = phosphate-buffered saline; GFAP = glial fibrillary acidic protein; DAPI = 4′,6-diamidino-2-phenylindole. Please click here to download this File.
