Before demonstrating the efficiency of this protocol in macrophages, nitrite, and BH4 levels were assessed in media supplemented with 10% of FBS containing either 10% of LCM or only recombinant M-CSF and GM-CSF. Nitrite, although considered for a long time as an end product of nitric oxide metabolism, is now regarded as physiological storage of nitric oxide, which can be recycled when required and thus is often used as an indicator of nitric oxide bioavailability7. BH4 is an essential cofactor of NOS for NO production. As seen in Figure 2, media supplemented with 10% FBS had similar nitrite levels regardless of M-CSF/GM-CSF or LCM. However, more variability between different LCM batches was observed. This observation also applied to the BH4 measurement. In fact, one batch of LCM (batch #3) had a significantly higher level of BH4 than the other two (batches #1 and #2). Furthermore, LCM batches contained significantly more biopterin than media supplemented with 10% FBS and recombinant cytokines. Significant variability of total biopterins between batches was also measured. These results demonstrate the importance of using a less variable source of M-CSF than LCM.
Moreover, media supplemented with 10% FBS contained significantly higher levels of nitrite compared with media containing either 2% or 5% of FBS. The difference between 10% and 5% was highly significant (p < 0.05) for BH4. However, similar levels of biopterins were quantified. In comparison, OptiMEM + 0.2% BSA was devoid of nitrite and BH4, making it a very clean and suitable media. However, as described by Bailey et al., although OptiMEM is meant to be used only once overnight when stimulating macrophages, cell death due to starvation was observed. DMEM: F12 supplemented with 2% of FBS was chosen to overcome this issue, allowing minimal nitrite and BH4 contamination while containing sufficient nutrients to obtain healthy cells. Indeed, despite some nitrite being detected, levels are negligible (~0.2 µM) compared to LPS/IFN stimulated WT macrophages (30-80 µM for 1 x 106 cells; data not shown).
To validate this improved protocol with experimental data, the isolation and characterization of BMDMs from a new BH4-deficient mouse model, the R26-CreERT2Gchfl/fl is presented here. BH4 is produced by the GTP cyclohydrolase I (Gch1) gene. As shown in Figure 3A, deficiency in Gch1 leads to loss of BH4 and the resulting disappearance of NO and subsequently nitrite, and an increase in superoxide anion causing oxidative stress as demonstrated previously by McNeill et al.8. Although this protocol has already been well defined and used to culture other BH4-deficient macrophages from different Cre-system such as the Gchfl/flT2C model6,8, the R26-CreERT2Gchfl/fl model is unique as it utilizes the conditional activation of the Cre-ERT2 system to remove Gch1 gene following tamoxifen treatment (Figure 3B,C). This allows knockout at various time points and the use of internal control in the form of a vehicle vs. tamoxifen treatment. In this example, cells were treated with either vehicle (95% Ethanol) or 0.1/1.0 µM 4-OH tamoxifen on days 1 and 3 (both vehicle and tamoxifen stock diluted 1:100 in media before 0.7/7.0 µL added to 1 mL of the existing culture volume).
As seen in Figure 3D, GTPCH protein is abolished following Tamoxifen treatment in the R26-CreERT2Gchfl/fl cells but not in the Gchfl/fl one. Importantly, both R26-CreERT2Gchfl/fl and Gchfl/fl control cells are still able to produce iNOS following LPS/IFN stimulation (Figure 3B,C). These changes in Gch1 gene expression and the resulting alteration in GTPCH protein led to significantly perturbed intracellular BH4 levels and attenuated production of nitrite. As shown in Figure 4, BMDMs isolated and cultured from R26-CreERT2Gchfl/fl mice exhibited significantly diminished BH4 production and decreased nitrite accumulation in the media, compared to those from control GCHfl/fl cells in the presence of tamoxifen. Similarly, the same cells cultured in LCM media also showed abolished Gch1 expression; although these cells still produced significant amounts of both BH4 and nitrite following knockout of Gch1 expression with 4 OH tamoxifen, possibly due to the contamination of media and FBS with BH4. This represents a clear improvement of the new method, over culturing the cells in L-cell-containing media.
Further characterization of this model demonstrates no significant differences in morphology between non-activated (M0) Gchfl/fl and R26-CreERT2Gchfl/fl BMDMs at Day 8 following different concentrations of tamoxifen. As shown in Figure 5, both models display a similar amount of adherent cells made of a round shape with elongated extremities, typically the features expected of unstimulated macrophages9.
Taken together, these results demonstrate that this method of isolation and culture of BMDMs provides a pure population of cells suitable for the culture of murine macrophages. This method allows for the culture of murine macrophages without the interference of exogenous compounds present in some media formulations.
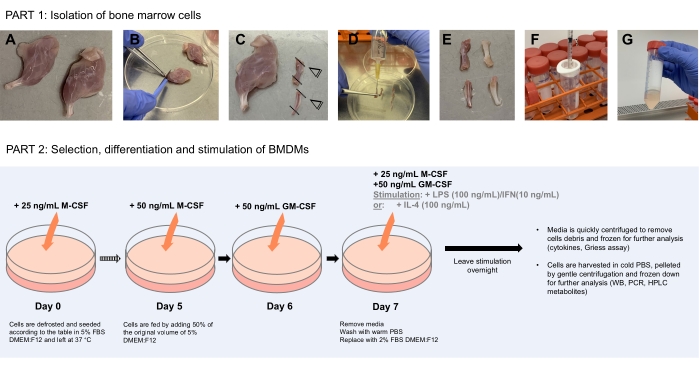
Figure 1: Schematic diagram illustrating bone marrow cells isolation (Part 1) to the selection, differentiation, and stimulation of BMDMs (Part 2). (A) Under sterile conditions, dissected legs are placed in a clean bacteriological petri dish after being washed in 70% ethanol. (B) Muscles are carefully removed using forceps and scalpel scraping along the bones. (C) The extremities of the bones are then cut off to expose the bone marrow. (D) The bone marrow is flushed from the bones using a 10 mL syringe filled with 10 mL of PBS by inserting the needle into the lumen of the bone. (E) The needle is run up and down through the bone to ensure all bone marrow is dislodged. The bone should appear white and clear at that stage with the dislodged bone marrow collected in the petri dish. (F–G) Repeat for all bones, collecting the disaggregated bone marrow in the 1 mL syringe, and pass it through a 70 µm cell strainer into a clean 50 mL centrifuge tube. Spin down and resuspend cells in freezing media for later use. Part 2 shows the selection, differentiation, and stimulation of BMDMs Please click here to view a larger version of this figure.
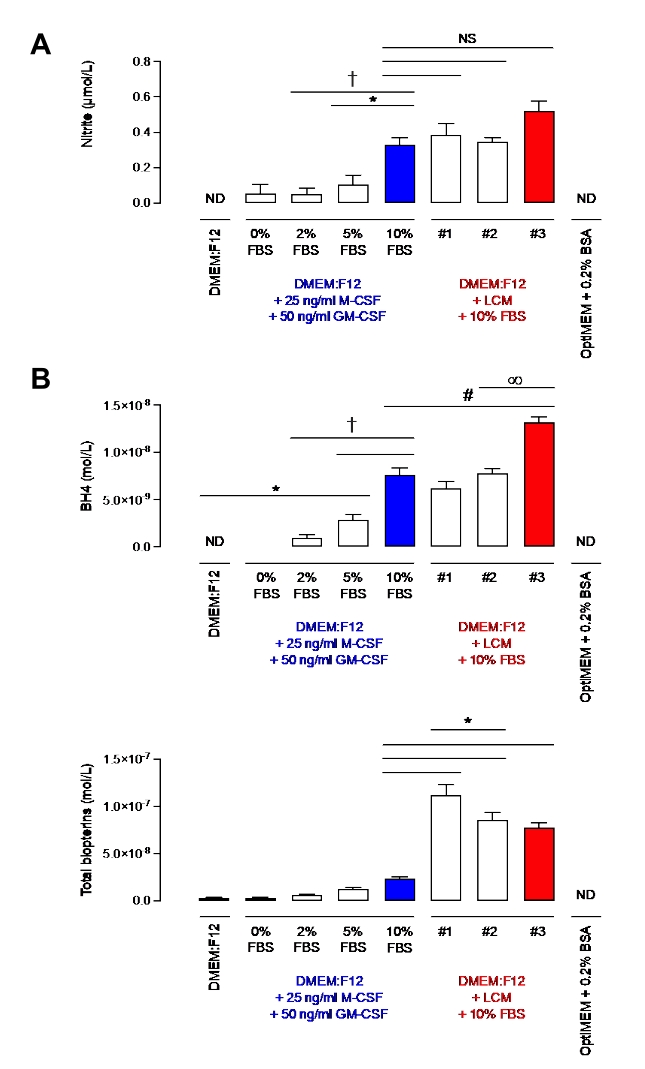
Figure 2: Nitrite and BH4 levels in DMEM: F12 + GM-CSF + M-CSF or DMEM: F12+ L-cell media. (A) Nitrite levels in different media were measured using the Griess Assay method. (B) BH4 and biopterins levels were measured in different media by HPLC quantification using a BH4 standard. Data are expressed as the mean (n = 3) ± SD. Data were analyzed using one-way ANOVA by multiple comparisons. Symbols represent p < 0.05 between groups. Please click here to view a larger version of this figure.
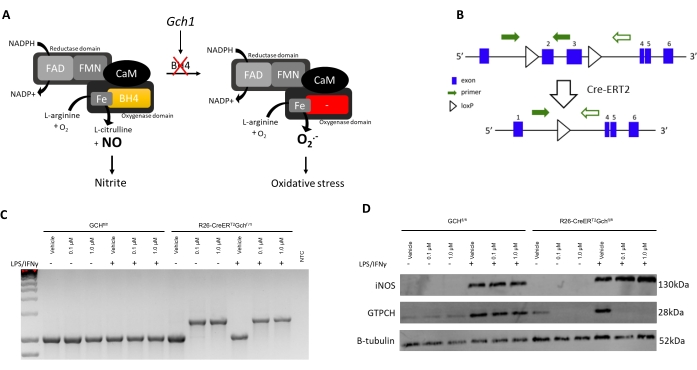
Figure 3: Introducing the R26CreERT2 model. (A) Scheme illustrating the role of BH4 as a NOS cofactor in redox biology. (B) Schematic detailing the excision of the critical exons (2 and 3) in Gch1 due to the flanking loxP sites and the conditional activation of Cre-ERT2 with tamoxifen in this murine model. (C) PCR (representative of n = 3 independent animals) demonstrating excision of the critical exon when BMDMs are treated with tamoxifen. WT mice produce a 1030 bp PCR product, while in the presence of the Cre, a 1392 bp product is produced, confirming excision of the critical exons. (D) Immunoblot of iNOS, GCH, and B-tubulin (representative of n = 3 independent animals). Please click here to view a larger version of this figure.
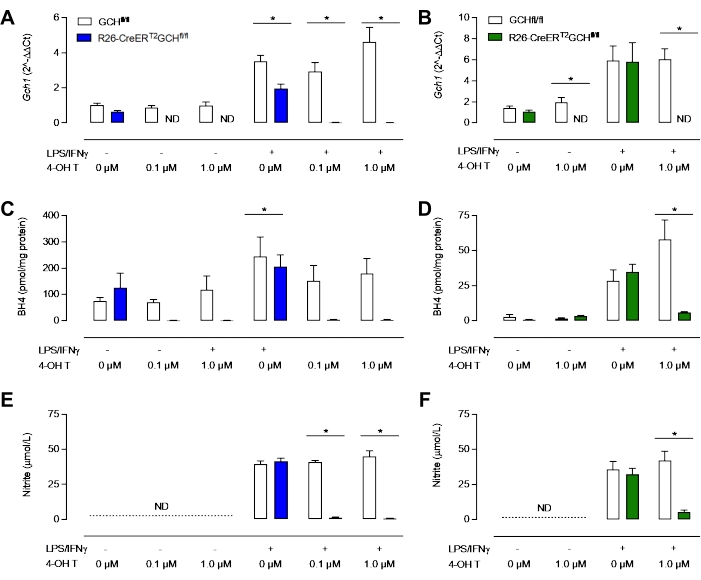
Figure 4: Characterization of BH4-deficiency in macrophages. (A,B) qRT-PCR confirms loss of Gch1 expression in the R26-CreERT2Gchfl/fl model treated with tamoxifen. Blue = New MCSF method, Green = classical LCM method. (C,D) Analysis of intracellular BH4 by HPLC reveals that 0.1 µM and 1.0 µM are sufficient to diminish BH4 levels significantly. (E, F) Measurement of nitrite in media using the Griess assay shows that unstimulated BMDMs do not produce detectable levels of nitrite, whereas Gchfl/fl and R26-CreERT2Gchfl/fl BMDMs produce nitrite in the presence of LPS/IFNγ. Significantly, tamoxifen treatment of R26-CreERT2Gchfl/fl significantly reduces nitrite levels in stimulated BMDMs. Data are expressed as the mean of (n = 3) ± SD. Data were analyzed using one-way ANOVA by multiple comparisons. Symbols represent p < 0.05 between groups. Please click here to view a larger version of this figure.

Figure 5: Comparison of BMDMs morphology. Photos obtained by optical microscopy of BMDMs at Day 8 with the varying treatment of tamoxifen. Scale is indicated in the image. Please click here to view a larger version of this figure.
| Culture Dish | Media Volume | Seeding Density |
| 12 well dish | 1 mL | 500,000 cells |
| 6 well dish | 2 mL | 1,000,000 cells |
| 100 mm dish | 10 mL | 3,000,000 cells |
| 75 cm2 flask | 15 mL | 5,000,000 cells |
Table 1: Representative seeding densities.
