General requirements for the protocol: protein purification facilities, UV-Vis spectrometer, high-field NMR spectrometer and operating software, post-processing analysis software including; NMRPipe32, Sparky33, (or CCPN Analysis34, or NMRViewJ35).
1. Recombinant expression and purification of a protein for PRE measurements
- Design an expression construct for the protein of interest so that there is a single cysteine residue present. Multiple mutations will be required to introduce a free cysteine at different positions in the protein of interest36.
- Express and purify a natural abundance (14N) or 15N-labeled sample of the protein of interest using an established protocol37.
NOTE: E. coli expression systems provide cost effective and robust method for recombinant protein expression since isotopic enrichment of 15N is a minimal requirement for biomolecular heteronuclear NMR spectroscopy. Typical steps are expression in minimal media, chromatographic purification, and removal of affinity purification tag. This protocol assumes a robust expression and purification protocol has been established that can produce sufficient protein of suitable quality for NMR investigations.- Maintain 1 mM reducing agent (DTT or TCEP) in buffers at all purification steps to prevent reaction of the free cysteine and formation of intermolecular disulfide bonds for IDPs.
NOTE: Some systems may be more tolerant and less aggregation prone to non-reducing conditions depending on the specific characteristics of the protein, as well as the temperature, pH, and buffer system chosen for purification38. - Remove affinity tags used for purification before proceeding since they may non-specifically interact with the protein in unpredictable ways or possibly contain reactive cysteine residues that could inadvertently serve as an unintended attachment site.
- Prepare a 15N labeled reference sample without cysteine mutation(s) mixed with a soluble version of the spin-label to assess the contribution of solvent PREs.
- Maintain 1 mM reducing agent (DTT or TCEP) in buffers at all purification steps to prevent reaction of the free cysteine and formation of intermolecular disulfide bonds for IDPs.
2. Conjugating the 3-Maleimido-PROXYL nitroxide spin label
- Store or exchange the purified protein into a degassed buffer containing 50 mM Tris pH 7 and 1 mM TCEP; the buffer may also contain up to 8 M urea if needed to aid protein solubility.
Alternatively, rapidly dilute a protein stock solution into at least 10 volume equivalents of degassed 50 mM Tris pH 7 and 1 mM TCEP buffer. Ensure that the protein concentration prior to adding spin-label is at least 100 µM. - Add 3-Maleimido proxyl from a stock solution to 20x molar excess of the protein of interest. Protect the sample from light and oxygen and incubate overnight at room temperature or 4 °C; gentle rocking or nutation may improve labeling efficiency.
- Prepare stock solutions of the spin-label by dissolving 3-Maleimido proxyl powder in 95% ethanol. Aliquots of the stock can be stored at -80 °C for less than 6 months.
- Critical step: Remove the non-reacted free spin-label to prevent non-specific solvent PREs. Achieve this by gel filtration or (preferably) extensive dialysis of the protein sample. This step will also introduce the protein into a buffer suitable for NMR.
NOTE: Reducing agents should be prepared fresh, and compatibility between buffer components should be considered; for example, TCEP degrades quickly in phosphate-based buffers, and this combination should be avoided39. - Treat all buffers used from this step forward with a chelating resin selective for divalent and transition metals to remove paramagnetic ions or spin-label quenchers. If the protein cannot be stored in an NMR buffer, concentrate the protein to be rapidly diluted into a buffer suitable for NMR.
- Monitoring the efficiency of spin-label incorporation.
- Use Ellman's reagent (5,5-dithio-bis-(2-nitrobenzoic acid) for quantifying free sulfhydryl groups in solution40.
NOTE: Detailed protocols are available from the manufacturer. For the purposes here, it is important to determine incorporation of the spin-label, the concentration of free sulfhydryl groups is compared with the total protein concentration. The percent of free sulfhydryl groups is the percent of molecules that do not have a nitroxide spin label attached. - Monitor the intensity of the peak corresponding to the tagged cysteine residue to judge spin-label incorporation into the protein of interest.
NOTE: This is a rapid and effective approach to determine the degree of spin labeling of the protein. Complete incorporation of the spin-label will result in the disappearance of the peak from the spectrum. With the poor dispersion characteristic of IDPs the peak corresponding to the mutant cysteine residue may not always be readily identified, and thus the use of Ellman's reagent (step 2.6.1) is recommended.
- Use Ellman's reagent (5,5-dithio-bis-(2-nitrobenzoic acid) for quantifying free sulfhydryl groups in solution40.
3. Prepare NMR sample for measuring intra- or inter-molecular PRE
- Prepare sample for measurement of intramolecular PRE
- Prepare 15N isotopically enriched, spin-labeled protein to a concentration of at least 100 µM but not more than 300 µM in a buffer suitable for NMR. Total sample volume (including D2O) is 500 – 550 µL.
NOTE: Common NMR buffers include phosphate, acetate, (bi)carbonate, and TRIS. Good's buffers such as MES, HEPES may also be appropriate. Exercise caution when selecting buffers to ensure no cross-reactivity with other solution components. - Ensure that the pH is ~7.2 or lower to minimize the effects of amide proton exchange with water. Keep the concentration of salt as low as possible (typically less than 150 mM), although the primary consideration is to maintain protein stability.
NOTE: Approaches for conducting NMR experiments in high-salt conditions have been described elsewhere41.
- Prepare 15N isotopically enriched, spin-labeled protein to a concentration of at least 100 µM but not more than 300 µM in a buffer suitable for NMR. Total sample volume (including D2O) is 500 – 550 µL.
- Prepare sample for measurement of intermolecular PRE
- Follow this step or step 3.1; they are not performed simultaneously. Prepare 14N natural abundance, spin-labeled protein in the chosen NMR buffer.
- Prepare the protein sample by mixing 15N isotopically enriched non-spin-labeled protein with 1%-50% 14N natural abundance spin-labeled protein so that the final concentration is identical to the sample prepared in 3.1.1. The total sample volume (including D2O) is 500 – 550 µL.
- Empirically optimize the ratio of the 15N and 14N proteins for each protein studied. The ratios of 1%, 5%, and 20% of 14N-spin-labeled protein are good starting points.
NOTE: A buildup of the PRE as a function of added 14N-spin-labeled protein indicates a specific effect; the observed PRE is sample-specific since it depends on distance and population (as discussed above), and therefore higher ratios of 14N-spin-labeled protein will be required if the interaction is particularly transient17.
- Transfer the NMR (either intra- or inter-molecular) sample to a 5 mm NMR tube that is appropriate for use in high-field magnets using a long-stem (9") glass pipette or micropipette. Ensure that all NMR samples include 5%-10% of D2O to facilitate field locking.
NOTE: NMR tubes that utilize polymer plugs to reduce the necessary sample volume are not recommended for PRE measurements due to difficulties related to effective sample shimming.
4. Set up NMR spectrometer and experiment specific parameters
- Exercise extreme caution when working around superconducting, high-field NMR spectrometers.
NOTE: Hazards include injuries due to the sudden acceleration of metallic objects toward the magnet, interference with implanted medical devices, and asphyxiation due to a sudden release of N2 and He2 gas in the event of a magnet quench. The following steps assume that the reader has undergone the required training, is aware of these and other local hazards, and has received approval from the facility manager to operate the NMR spectrometer. When in doubt of a step or instruction, consult with the facility manager or experienced user to prevent potential personal injury or damage to the spectrometer. - The following steps assume a commercial NMR spectrometer running a modern version of the acquisition control software. Download the pulse program and parameter files and place them in the appropriate directories.
NOTE: A pulse program and parameter set suitable for use with a Bruker spectrometer and TopSpin (3.2 or later) are available upon request from the authors.- Critical step: Familiarity with installing non-native NMR pulse programs is assumed; consult with the facility manager or an experienced user if necessary.
- Place the sample in the magnet, lock on the 2H signal using the Lock command, tune and match the 1H channel according to facility protocols (the exact procedure will depend on if the probe is equipped with a remote tune and match module).
- Adjust the shims using the topshim subroutine to optimize solvent signal suppression.
- Calibrate the 1H and 15N 90° pulses using standard methods.
- Calibrate the 1H pulse using the popt program (use pulsecal first to estimate pulse length).
- Calibrate the 15N pulse against a standard sample; make sure this value has been calibrated recently by discussing with a technical director or experienced user.
- Alternatively, calibrate the 15N pulse on the sample by varying one of the 90° pulses of an HMQC experiment until a null signal is achieved.
- Determine the correct attenuation for shaped pulses using the shape tool (stdisp) subroutine.
- Open the appropriate pulse shape file by clicking on the folder icon. The shaped pulses are found in the pulse parameters section of ACQUPARS.
- Load the pulse definition file and click on Analyze Waveform > Integrate Shape. Input the calibrated 1H 90° hard pulse, desired shaped pulse length, and rotation (90° or 180°).
- Calculate the power level of the shaped pulse by adding the change of power level to the attenuation for the calibrated 90° pulse.
- Record a standard 1H, 15N HSQC (hsqcetfpf3gpsi) to optimize sweep width, carrier frequency and check water suppression25.
- Adjust the sweep width and the number of indirect dimension increments using the sw and td commands or directly in the appropriate dialog boxes. Typically, for collecting PREs, spectral widths are chosen so that the spectrum is not folded.
5. Setup the 1HN–T2 experiment
- Calibrate the shaped pulses as described above (4.4.5-4.5.7). The shaped pulse parameter files for the PRE experiment are Eburp2.1000 (90° pulse), Reburp.1000, and Iburp2.1000. Enter the calibrated pulse lengths in the pulse parameters section on the ACQUPARS tab.
- This experiment measures the 1HN–T2 using the two time-delay point approach30.
- Set the time delays by editing the vdlist file, the first delay (Ta) is set to 0.01 ms.
- Choose the second delay, (Tb) using the relationship to the expected maximum PRE (Tb ~ 1.15/(R2,dia + Γ2) where R2,dia represents the R2 of the diamagnetic sample13. Without prior knowledge of the magnitude of the PRE contribution to the observed relaxation, a good starting point is setting Tb to ~1x 1H T2.
- Then determine a suitable value by comparing the first increments (processed with the efp command) of the Ta and Tb spectra and adjusting Tb such that the signal decays to between 40%-50% of its initial value.
NOTE: This approach optimizes the spectral signal-to-noise, a necessary consideration for samples that cannot be highly concentrated (< 50 µM). Suitable values of Tb are sample dependent but typically range from 8 – 40 ms for an average sized protein.
- Determine the number of complex points to record and number of scans for sufficient signal averaging. Since IDPs have longer 15N T2 than folded proteins of comparable size, longer acquisition times in the indirect dimension may be used.
NOTE: This value is dependent on the specific characteristics of the protein but can be roughly estimated from the 15N T2 and optimized by monitoring signal decay in the FID. For the direct dimension, 1024* complex points (13 ppm sweep width, 112.6 ms acquisition time) is sufficient for most samples. - Use the command expt to calculate the experiment time and then start the experiment with the command zg.
6. Make a diamagnetic sample by reducing spin-label with ascorbic acid
- Dissolve sodium ascorbate in the NMR buffer and adjust the pH to match the original NMR buffer.
- Calculate the concentration of sodium ascorbate stock so that a 10x molar excess of ascorbate over the concentration of the spin-label can be added with the least change of sample volume. For example, for a 100 µM protein sample, a 100 mM stock of ascorbate is appropriate. Reducing the spin-label will require adding 5.5 µL of ascorbic acid stock solution, which is only 1 % of the total sample volume.
- Add the required amount of ascorbic acid to the NMR tube by placing a droplet below the rim of the tube, cap the tube, carefully invert the tube to mix, and then spin at 200-400 x g for 10-20 s in a hand-cranked centrifuge to settle the sample at the bottom of the tube.
- Wrap the NMR tube in foil to protect from light and allow the reaction to proceed for at least 3 h.
- Record 1HN–T2 on the diamagnetic sample using the same parameters used for the paramagnetic sample.
- Recalibrate the pulses. However, they should not have changed from the paramagnetic measurements; if they are significantly different (> 0.5 µs difference), consider sample quality (e.g., degradation, precipitation).
- Ensure that all acquisition parameters, including the specified relaxation delays (vdlist), number of dummy scans, number of scans collected, number of complex points collected, acquisition time, sweep widths, and carrier frequencies remain the same for the diamagnetic and paramagnetic samples.
7. Process paramagnetic and diamagnetic spectra
- Copy the data to local computer or workstation that has NMRPipe and Sparky installed and configured. Make a folder named proc in the experiment data directory that contains the ser file.
- Copy the NMRPipe scripts fid.com, p3d.com, and nmrproc.com into proc (processing scripts are available upon request from the authors).
- Use the fid.com script to convert the Bruker data format (ser) into NMRPipe format.
- Use the p3D.com script to split the pseudo3D planes into individual spectra.
- Use the nmrproc.com script to read the output of the fid.com script, apply solvent suppression, a window function, append zeros to the raw data (zero fill), apply phase correction, execute a Fourier transformation, trim the data for display and write the processed data to disk. The script will output one file for each relaxation delay recorded (Ta and Tb).
NOTE: Each of these scripts is customizable to optimize the processing for the specific details of each experiment. Tutorials and example data sets are included in the NMRPipe distribution available from the NMRPipe website32. NMRDraw may be used for spectral viewing during processing (e.g., setting proper phase angles etc.). Options available for NMRPipe commands can be viewed using the command nmrPipe -help.
8. Transfer resonance assignments and extract peak heights
- Change the file header information for each spectrum file (Ta, Tb for both paramagnetic and diamagnetic samples) using the command sethdr [filename] -ndim 2.
- Use Sparky to extract peak heights33 following steps 8.3-8.5. Other software packages, including NMRPipe (NMRDraw)32, CCPN Analysis34, and NMRViewJ35 are also appropriate.
- Read the spectral files into Sparky. At this step the data set will consist of one spectrum for each time point spectra (Ta, Tb), for both the paramagnetic and diamagnetic samples, measured for each position of the spin-label in the protein.
- Use Sparky to pick peaks (command: F8, then click and drag) and transfer assignments using the transfer peak list tool from a reference peaklist.
NOTE: Resonance assignments of the protein of interest are necessary for sequence-specific interpretation of the observed PREs36.- Set contours in both paramagnetic and diamagnetic spectra to the same level. Ensure to set the contours so that the spectra collected after the time delay do not purposely exclude peaks but are high enough so that the Ta spectra are not overly noisy.
- Save the new peak lists for each spectrum and include the measured peak intensity and Sparky calculated signal to noise ratio (command: lt to open peaklist, click options to include intensity and SNR columns, command: save).
9. Extract 1HN–T2 rates for each residue and calculate PRE
- Import the peak lists into spreadsheet software or a preferred programming language such as Python.
NOTE: For each spin-label position on the protein, the dataset will consist of four peak lists with associated peak intensities, one each of Ta and Tb for both the paramagnetic and diamagnetic experiments. - Calculate 1HN R2 for both the paramagnetic and diamagnetic samples using the equation:
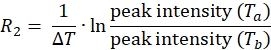
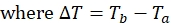
- Use the above equation to determine the relaxation rate for each residue for the paramagnetic and diamagnetic samples.
- Determine the 1HN–Γ2 rate for each residue using the equation:
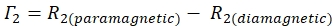
- Use the Sparky calculated signal to noise ratio (SNR) to compute the uncertainty of the peak height for each residue.
- Propagate the error using the equation:
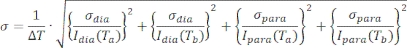
- Plot 1HN–Γ2 as a function of residue number using a scatter plot including the error calculated in 9.6.
Intramolecular 1HN–Γ2 PREs were recorded on a self-associating, intrinsically disordered fragment (residues 171-264) derived from the low-complexity domain of the RNA-binding protein EWSR142 (Figure 3). Residues in close sequential proximity to the spin-label attachment point (e.g., residue 178 or 260 in Figure 3) are expected to be significantly broadened and are not detectable in the spectrum. Residues sequentially spaced from the attachment point yet show enhanced Γ2 were spatially close (10-35 Å) to the spin-label. In the case of EWSR1 171-264, attributing the source of the PRE effect is complicated since it may arise from a combination of inter- and intra- residue contacts and is dependent on the distance from the nucleus to the paramagnetic center, the population of that conformation, and the dynamics of the vector connecting the electron and nuclear spins. Further, the magnitude of PREs arising from intramolecular contacts is not concentration-dependent, while PREs arising from intermolecular contacts depend on concentration as well as the kinetics and dynamics of the association between protein molecules.
A possible interpretation of these data is that the IDP ensemble samples conformations that are more compact than an extended chain. Alternatively, the PREs could arise from intermolecular contacts responsible for the self-association of EWSR1, or the PREs could be from a combination of both intra- and intermolecular contacts. In the case presented here, what remains unknown is how close the residues approach the spin-label or for how long they remain in close proximity. With highly flexible molecules such as EWSR1 171-264, it can be difficult to qualitatively disentangle these parameters. By placing the spin-label at different residue positions, contacts between different parts of the chain may be identified, providing a more accurate interpretation of specific interactions that may be functionally relevant for self-association (Figure 3). Measuring intermolecular PREs (14N spin-labeled protein mixed with 15N non-spin labeled protein), employing a mutational strategy of residues with larger than average PREs (e.g., residues 196 or 215, Figure 3), and utilizing other biophysical methods such as dynamic light scattering, size exclusion chromatography, and analytical ultracentrifugation, are useful for characterizing the conformational ensemble of an IDP.
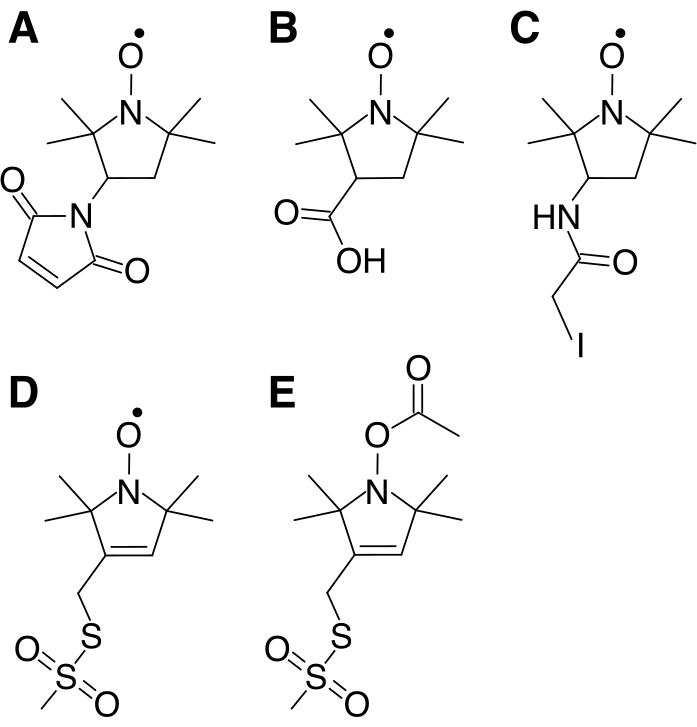
Figure 1: Molecules containing an unpaired electron and various functional groups to facilitate attachment to free cysteine residues that are typically used as paramagnetic relaxation agents. Diamagnetic molecules may be used as controls. (A) 3-Maleimido-2,2,5,5-tetramethyl-1-pyrrolidinyloxy, free radical (3-Maleimido-PROXYL) (B) 3-Carboxy-2,2,5,5-tetramethyl-1-pyrrolidinyloxy, free radical (3-Carboxy-PROXYL) (C) 3-(2-Iodoacetamido)-2,2,5,5-tetramethyl-1-pyrrolidinyloxy, free radical (3-(2 – Iodoacetamido-PROXYL) (D) 1-Oxyl-2,2,5,5-tetramethylpyrrolidin-3-yl) Methyl Methanethiosulfonate (MTSL) (E) (1-Acetoxy-2,2,5,5-tetramethyl-δ-3-pyrroline-3-methyl) Methanethiosulfonate (Acetoxy-MTSL) Please click here to view a larger version of this figure.
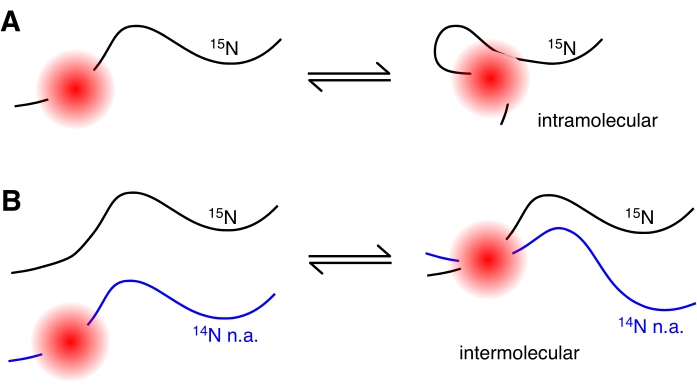
Figure 2: Depiction of intra- and intermolecular PRE. (A) Intramolecular PRE, the red circle represents the effective radius of a paramagnetic center attached to a 15N-labeled protein. The PRE effect decreases with an <r-6> dependence on distance from the paramagnetic molecule. (B) Intermolecular PRE, the paramagnetic group (red circle), is located on a 14N (natural abundance) protein (blue) that is invisible to NMR. The effects of the paramagnetic group on the non-NMR active protein are observed as increased relaxation rates when it comes into close contact with the 15N protein (black). Please click here to view a larger version of this figure.
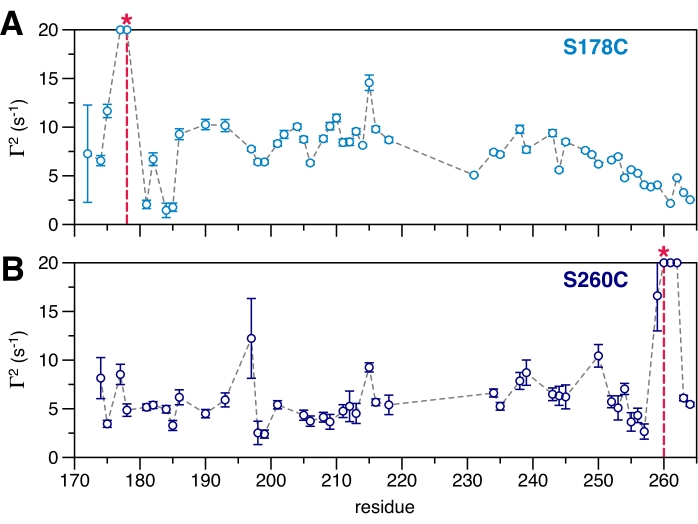
Figure 3: 1HN–Γ2 rates for residues 171-264 of the intrinsically disordered domain of EWSR1. A serine residue at position (A) 178 or (B) 260 that has been mutated to a cysteine serves as the attachment point for a 3-Maleimido-PROXYL spin-label (red *). Increased relaxation rates occur at the location of the tag, other sites of increased relaxation are indicative of intramolecular interactions. Please click here to view a larger version of this figure.
