For analyzing microbiome bacteria, specific and universal FISH probes to bacterial 16S were utilized on wild-isolated animals. Wild Caenorhabditis tropicalis strain (JU1848) was sampled from the Nouragues forest near a small river in the French Guiana from rotting palm tree fruits22. Under the differential interference contrast (DIC) microscope, this nematode strain was found to be colonized with a bacterium that appears to directionally adhere to the intestinal epithelium (Figure 2A). JU1848 was then selectively cleaned to eliminate other microbial contaminants and enrich for the desired adhering bacterium23. Using the universal PCR method, the bacterium was identified as a new species in the Alphaproteobacteria class. A FISH probe labeled with Cal Fluor Red 610 was then designed specifically to the 16S rRNA sequence of this bacterium to allow fluorescent visualization of colonization within C. tropicalis (Figure 2B). A universal 16S rRNA FISH probe capable of binding many species of bacteria (EUB338) was labeled with 6-carboxyfluorescin (FAM) and was also added to this sample. The green and red fluorescent signals overlap completely, suggesting that most bacteria colonizing the intestines are the adhering Alphaproteobacteria bacterium. These animals were fixed in PFA before staining.
For analyzing experimental infection in the lab with intracellular pathogens of known identity, Orsay virus and microsporidian-specific FISH probes were utilized on C. elegans with a wild-type background. The Orsay virus is a positive-strand RNA virus from the Nodaviridae family, and the only natural viral pathogen found in C. elegans. The bipartite RNA genome of the Orsay virus consists of RNA1 and RNA2 segments, and FISH probes targeting both of these segments have been developed (Figure 3A,B)9,18. In the intestine, viral RNA is sensed by RIG-I homolog DRH-124, which is required for activation of the transcriptional defense program named the Intracellular Pathogen Response (IPR)25,26,27. The transcription of antiviral IPR genes is at least partially controlled by the ZIP-1 transcription factor21. Here, the expression of ZIP-1::GFP is seen localized in the intestinal nuclei of cells that show positive Orsay virus FISH staining in the cytoplasm (Figure 3A)21. Multiple animals stained with Orsay-specific FISH are shown to indicate the strength of this signal for easy quantification (Figure 3B). Animals shown in Figure 3A,B were fixed in PFA.
The microsporidian parasite named Nematocida parisii, meaning nematode-killer from Paris, is an obligate intracellular pathogen of the intestine. Several FISH probes that label 18S rRNA of N. parisii have been used, including fluorescently tagged MicroA and MicroB probes. Multiple animals stained with MicroB FISH are shown to indicate the strength of this signal for easy quantification (Figure 3C). Additionally, C. elegans is infected by other closely related microsporidia. Co-infection of N2 with N. parisii and the related N. ausubeli can be distinguished using this FISH protocol by designing species-specific FISH probes that compete against each other for binding to a divergent region on the 18S rRNA (Figure 3D)28. In this example, the N. parisii FISH probe has perfect base-pairing to the N. parisii 18S rRNA, but a 7 bp mismatch to N. ausubeli 18S rRNA. The converse is true for the N. ausubeli probe. As such, each species-specific FISH probe will outcompete for binding to the cognate species 18S over the non-cognate species. Additionally, the use of DAPI to stain nuclei allows for better localization of the infection in the context of the whole animal, especially for the intestine which has large, easily identifiable nuclei. Figure 3C,D contains animals that were fixed in PFA. Later infections with N. parisii result in the development of meronts into spores. To visualize N. parisii spores, the animals must be fixed in acetone as it penetrates the spore wall better than PFA (Figure 3E,F)8. The resulting FISH staining demonstrates the small and large rod-shaped structures, that likely correspond with N. parisii spores, which are stained with N. parisii-specific probes in red.
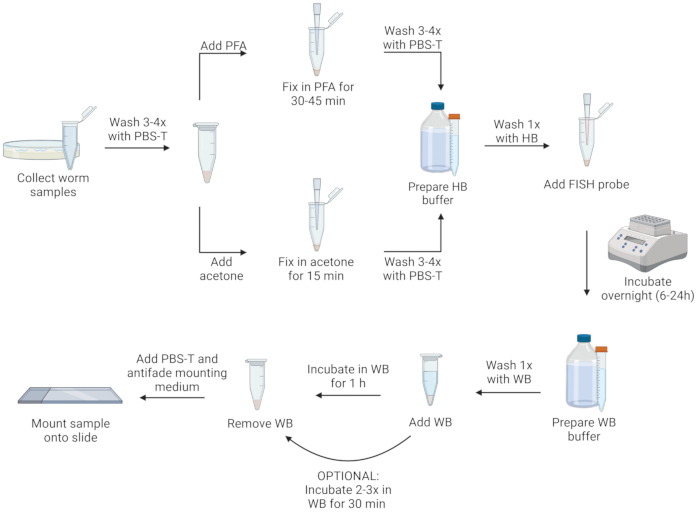
Figure 1: Visual representation of FISH protocol. Created with Biorender.com. Please click here to view a larger version of this figure.
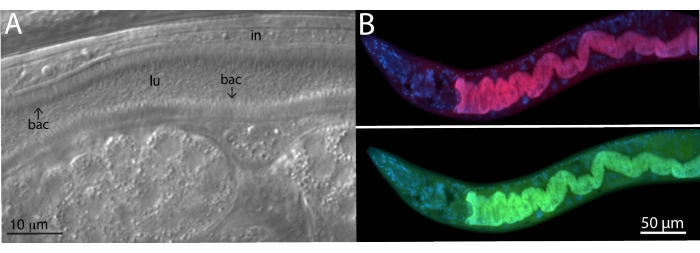
Figure 2: FISH staining of wild C. tropicalis JU1848 strain colonized with adhering bacteria in the intestines. (A) Nomarski image depicting thousands of thin bacilli bacteria (bac) directionally binding to the intestine (in) of JU1848, creating a hair-like phenotype within the lumen (lu). This figure panel is adapted from Morgan, E. et al. (2021)23. (B) FISH staining of JU1848, fixed in PFA, using a red labeled probe (b002_16S_A-CF610) designed to target the 16S rRNA sequence of the adhering bacterium (top) and a green-labeled universal FISH probe (EUB338-FAM) designed to target the 16S of bacteria (bottom). DAPI staining of host nuclei is shown in blue. See Table 1 for probe sequences. Please click here to view a larger version of this figure.
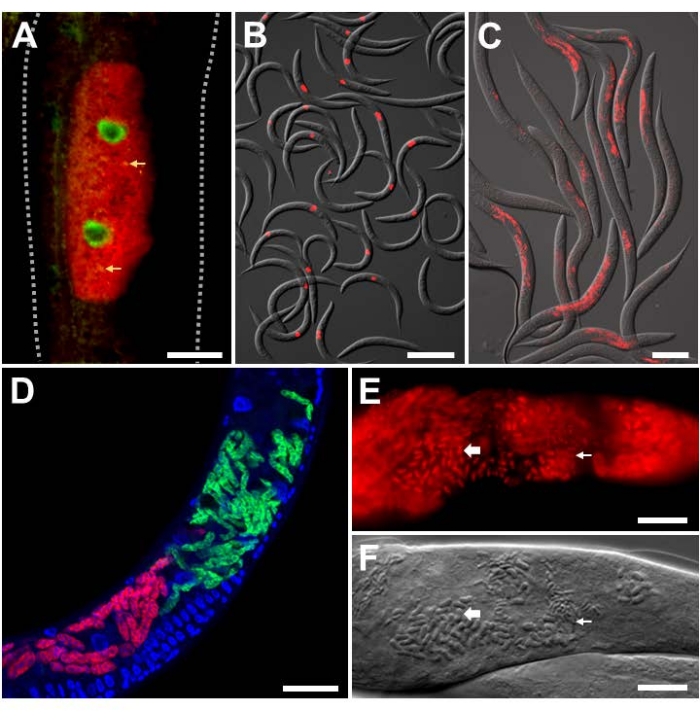
Figure 3: FISH staining of C. elegans infected with intracellular pathogens. (A,B) FISH staining of C. elegans expressing ZIP-1::GFP and infected with the Orsay virus, which were fixed with PFA before staining to preserve the GFP signal. Orsay 1 Red and Orsay 2 Red probes were used for pathogen staining. (A) The composite image consists of merged red and green fluorescent channels. Nuclear ZIP-1::GFP expression is induced upon Orsay virus infection and is shown in green. Autofluorescence from the gut granules is shown in yellow and indicated with yellow arrows. Dotted lines outline the nematode body. Scale bar = 25 µm. (B) The composite image consists of merged red fluorescent and DIC channels. Scale bar = 200 µm. (C,D) FISH staining of wild-type C. elegans infected with microsporidia that were fixed in PFA. (C) FISH staining of wild-type C. elegans infected with N. parisii and fixed in PFA. MicroB-CF610 probe was used for pathogen staining. The composite image consists of merged red fluorescent and DIC channels. Scale bar = 100 µm. (D) FISH staining of wild-type C. elegans co-infected with N. parisii and N. ausubeli in the intestine. The two pathogens were co-stained using a pair of specific FISH probes that compete for binding to the same region of the 18S rRNA. N. parisii was stained using MicroF-CF610 (red) and N. ausubeli was stained using MicroSp1A-FAM (green). DAPI staining of host nuclei is seen in blue. Scale bar = 25 µm. (E) FISH staining with acetone-fixed wild-type C. elegans infected with N. parisii spores. MicroA-CF610 (red) was used for staining (red). Scale bar = 15 µm. (F) Nomarski image depicting N. parisii spores seen in (E). Scale bar = 15 µm. In (E) and (F), the small and large rod-shaped structures are labeled with small and large arrows, respectively, that correspond to N. parisii spores. See Table 1 for probe sequences. The image shown in (A) is adapted from Lažetić, V. et al. (2022)21. Images shown in (B) and (C) are adapted from Reddy, K. C. et al. (2019)26. Images shown in (E) and (F) are adapted from Troemel, E. R. et al. (2008)8. Please click here to view a larger version of this figure.
| Probe name | Probe specificity | Probe fluorophore | Probe sequence |
| EUB338-FAM | Bacterial 16S (universal) | 5' 6-fluorescein (FAM) | GCTGCCTCCCGTAGGAGT |
| b002_16S_A-CF610 | Alphaproteobacteria 16S | Cal Fluor Red 610 (CF610) | TGTACCGACCCTTAACGTTC |
| Orsay1 Red | Orsay virus RNA1 | Cal Fluor Red 610 (CF610) | GACATATGTGATGCCGAGAC |
| Orsay2 Red | Orsay virus RNA2 | Cal Fluor Red 610 (CF610) | GTAGTGTCATTGTAGGCAGC |
| MicroA-CF610 | Nematocida parisii 18S | Cal Fluor Red 610 (CF610) | CTCTGTCCATCCTCGGCAA |
| MicroB-CF610 | Nematocida parisii 18S | Cal Fluor Red 610 (CF610) | CTCTCGGCACTCCTTCCTG |
| MicroF-CF610 | Nematocida parisii 18S | Cal Fluor Red 610 (CF610) | AGACAAATCAGTCCACGAATT |
| MicroSp1A-FAM | Nematocida ausubeli 18S | 5' 6-fluorescein (FAM) | CAGGTCACCCCACGTGCT |
Table 1: List of FISH probe sequences. All FISH probes were commercially purchased with the fluorophore attached to the 5' end (via custom oligonucleotide synthesis; see Table of Materials) and the oligonucleotides were purified by reverse-phase HPLC.
