This protocol provides step-by-step instructions for the caspase protein induction in E. coli, purification of the recombinant Drosophila caspases Dronc and Drice, the synthesis of candidate substrates, and the in vitro cleavage reaction with candidate substrates (here DriceC211A) and the caspase Dronc. The catalytic mutant DriceC211A was used as model substrate in this assay because it does not have auto-processing activity (Figure 2A) and remains full-length until it is cleaved by Droncwt. DriceC211A should always be used as a positive control to validate that the Droncwt preparation has enzymatic activity.
Figure 2A provides a representative example of the expression and purification of recombinant caspases. Four different recombinant caspases were induced and purified: 6xHis-Droncwt, 6xHis-DroncC318A, 6xHis-Dricewt, and 6xHis-DriceC211A. The purified caspases were run by SDS-PAGE, immunoblotted, and the blot was probed with an anti-His antibody (diluted 1:5,000; followed by anti-mouse IgG, HRP-linked antibody (1:10,000)). The unprocessed 6xHis-Dronc (proDronc) runs at a relative molecular weight (MWr) of 55 kDa (lane 2) and unprocessed 6xHis-Drice (proDrice) has a MWr of 35 kDa (lane 4). Auto-processing of the caspases is visible by the appearance of bands of smaller MWr which due to the presence of the His-tag at the N-terminus represent the large subunits of the caspases (Figure 1). In the case of 6x-Dronc, the large subunit has a MWr of 40 kDa (lane 1). The large subunit of 6x-Drice runs at 23 kDa (lane 3). The catalytic mutants 6xHis-DroncC318A and 6xHis-DriceC211A fail to auto-process and are only detectable as full-length proteins (lanes 2 and 4).
Figure 2B To demonstrate that the bacterially produced and purified 6xHis-Droncwt preparation has enzymatic activity, an in vitro cleavage assay as described in this protocol was performed. As negative control, the catalytic mutant 6xHis-DroncC318A was used. The substrate was RRL-generated N-Myc-DriceC211A, which is tagged with a Myc-tag at the N-terminus. After the in vitro cleavage reaction, the proteins were separated by SDS-PAGE, immunoblotted and the blots were incubated with an anti-Myc antibody (diluted 1:1,000) followed by anti-mouse IgG, HRP-linked antibody (1:10,000)) to detect N-Myc-DriceC211A. Successful cleavage and thus enzymatic activity of the caspase can be demonstrated by the appearance of at least one band of smaller MWr compared to the full-length, unprocessed form of the substrate. Unprocessed, full-length N-Myc-DriceC211A has a MWr of 40 kDa (lanes 2 and 4), whereas the large subunit of processed N-Myc-DriceC211A runs at 30 kDa (lane 3). Lane 1 represents the unprogrammed RRL lysate (no plasmid/transcript added). Lane 2 demonstrates in vitro production of DriceC211A by RRL expression. Lane 3 contains the in vitro cleavage reaction with 6xHis-Droncwt. Lane 4 contains the in vitro cleavage reaction with 6xHis-DroncC318A.
Figure 2C An in vitro cleavage reaction that uses both recombinant and purified caspase (6xHis-Droncwt and 6x-His-DroncC318A) and substrate (6xHis-DriceC211A) according to this protocol, was analyzed by SDS-PAGE and immunoblot. For analysis of the cleavage reaction, the anti-cleaved Drice antibody (diluted 1:5,000; followed by anti-rabbit IgG, HRP-linked antibody (1:10,000)) was used in this immunoblot. The anti-cleaved Drice antibody detects its neo-epitope in the large subunit (23 kDa) of 6xHis-DriceC211A only after processing by 6xHis-Droncwt (lane 1). The catalytic mutant 6xHis-DroncC318A is unable to process 6xHis-DriceC211A in this assay and full-length 6xHis-DriceC211A appears as faint unprocessed band of 35 kDa (lane 2).
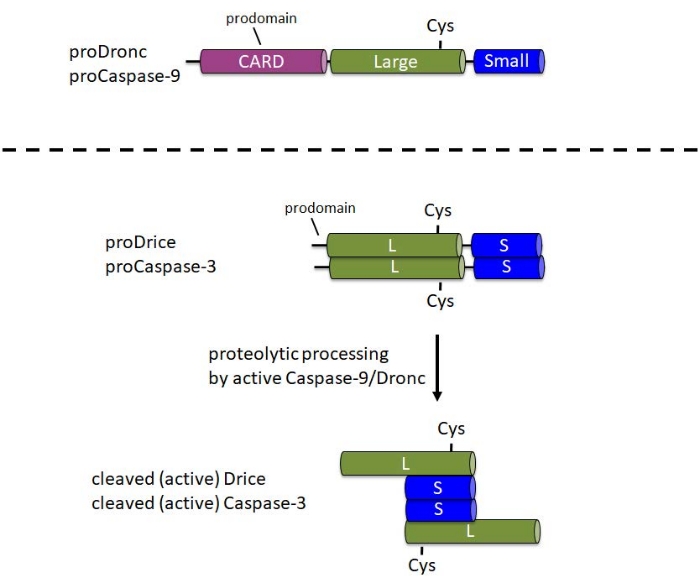
Figure 1: Domain structure of initiator caspases Caspase-9 and Dronc and effector caspases Caspase-3 and Drice. CARD – caspase activation and recruitment domain; Cys – relative location of the catalytic Cysteine residue; L – large subunit; S – small subunit. The location of the N-terminal prodomains is indicated. Please click here to view a larger version of this figure.
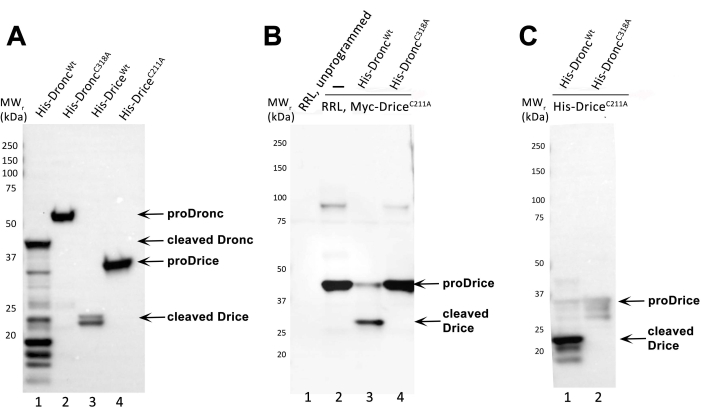
Figure 2: Representative results. (A) Immunoblot analysis of purified recombinant 6xHis-Dronc and 6xHis-Drice preparations, probed with an anti-His antibody. Unprocessed (proDronc and proDrice) and cleaved 6xHis-Dronc and 6xHis-Drice are indicated by arrows. MW markers are indicated on the left. (B) Immunoblot analysis of the in vitro cleavage reaction of RRL-generated N-Myc-Drice with caspases 6xHis-Droncwt (lane 3) or 6x-His-DroncC318A (lane 4) probed with an anti-Myc antibody. Unprogrammed and programmed (N-Myc-DriceC211A) RRL reactions are loaded and separated in lanes 1 and 2. Unprocessed (proDrice) and cleaved N-Myc-Drice are indicated by arrows. MW markers are indicated on the left. (C) Immunoblot analysis of the in vitro cleavage reaction of bacterially expressed and purified recombinant 6xHis-DriceC211A with caspases 6xHis-Droncwt (lane 1) or 6x-His-DroncC318A (lane 2), probed with an anti-cleaved Drice antibody. Full-length (proDrice) and cleaved 6xHis-DriceC211A are indicated by arrows. MW markers are indicated on the left. Please click here to view a larger version of this figure.
Supplementary Table 1: This table contains primers used for cloning in pET28a vector. Please click here to download this File.
Supplementary Table 2: This table contains the composition of buffers and media. Please click here to download this File.
