1. Preparation of reagents
- Mueller-Hinton broth (MHB) preparation: Dissolve 22 g of cation-adjusted MHB in 1 L of deionized water. Autoclave the media at 121 °C and 15 lb pressure for 20 min, making sure the bottle is loosely capped. Store the prepared sterile broth at 4 °C until use.
- Tryptic soy agar (TSA) preparation: Dissolve 20 g of TSA powder in 500 mL of deionized water. Autoclave the agar mixture at 121 °C and 15 lb for 20 min. Transfer the sterile media to a 55 °C water bath to bring down the temperature of the molten agar. Pour the cooled prepared agar onto sterile Petri dishes (~15 mL), and allow it to solidify. Store the solidified agar plates inverted to avoid condensation.
- Tryptic soy broth (TSB) preparation: Dissolve 15 g of TSB powder in 500 mL of deionized water. Autoclave the broth mixture at 121 °C and 15 lb for 20 min. Store the prepared sterile broth at 4 °C until use.
- Phosphate-buffered saline (PBS): Autoclave commercially purchased 1x PBS at 121 °C and 15 lb for 20 min before sterile use.
- Microbial viability assay reagent preparation: Equilibrate the contents of the assay to room temperature. Mix 10 mL of buffer with the lyophilized luminescent reagent powder, and store the prepared reagent at −20 °C as 1 mL aliquots. Before each time point, thaw the light-sensitive reagent, and bring it to room temperature.
- Gentamicin sulfate stock solution: Dissolve 80 mg of gentamicin sulfate (GS) in 10 mL of 1x PBS. Vortex the drug solution thoroughly to obtain an 8 mg/mL GS stock solution.
- Vancomycin hydrochloride stock solution: Dissolve 10 mg of vancomycin hydrochloride powder thoroughly in 10 mL of deionized water to obtain a 1 mg/mL stock solution.
- Boric acid buffer solution: Dissolve 24.7 g (0.4 M) of boric acid in 900 mL of deionized water. Adjust the pH of the solution to 10.4 with a 50% (w/v) sodium hydroxide solution. Further to this step, add deionized water to make it to 1 L.
- O-pthaldialdehyde reagent (OPA): Dissolve 0.2 g of OPA in 1 mL of methanol. Add 19 mL of 0.4 M boric acid buffer (pH 10.4) to the OPA solution. Add 0.4 mL of 2-mercaptoethanol to the OPA-boric acid mixture. Cover the reagent vial in aluminum foil, and store at 4 °C for use up to 2-3 days post preparation.
CAUTION: Avoid contact with the skin, eyes, and clothing. Handling of the OPA and 2 mercaptoethanol reagents should be performed only under a fume hood with appropriate exhaust ventilation while wearing proper personal protective equipment. Avoid breathing in the vapors, dust, or aerosols.
2. Preparation of virgin and drug-loaded UHMWPE
- Sift 2 g each of gentamicin sulfate and vancomycin hydrochloride powders with a sieve (75 µm pore size), and dehydrate at 45 °C in a vacuum oven (<0.1 atm) for 18-24 h.
- Blend the dehydrated drug with UHMWPE powder at 7% w/w (1.12 g of antibiotic + 14.88 g of UHMWPE) in a container using a mechanical mixer for 5 min.
- Preheat an aluminum bronze custom mold (85 mm x 50 mm) with stainless steel insert plates to 180 °C in a convection oven for 1 h. Preheat the platens of the press to 170 °C.
- Add the blended powder to the mold, and compress at 10 MPa, 170 °C for 10 min, followed by a 45 min water cooling cycle at 10 MPa.
- Remove the molded UHMWPE block (~3.5 mm) from the press using a hydraulic press.
- Mill the top and bottom surfaces of the block using a computerized numerical control (CNC) to remove any surface irregularities and contaminants.
- Cut the block into 3 mm x 5 mm x 20 mm strips using the CNC.
- Prepare virgin UHMWPE (without the addition of antibiotics) using the same methodology as described as a control for the study.
Supplementary Figure 1: Parts of the mold used for molding the UHMWPE samples. (A) Stainless steel insert plate; (B) mold cavity; (C) plunger; (D) backplate Please click here to download this File.
3. Determining the elution kinetics of drug-eluting UHMWPE
- Place a 3 mm x 5 mm x 20 mm strip in a 3 mL syringe with a Luer lock and a 25 G needle.
- Wash the strip by filling the syringe with deionized water and inverting the syringe multiple times.
- Fill the syringe with deionized water up to the 2 mL graduation.
- Place the syringe setup on a shaker at 100 rpm at room temperature.
- At each time point (6 h, 1 day, 2 days, 3 days, 1 week), collect the eluent in a 2 mL vial.
- At each time point, wash the strip by filling the syringe with deionized water and inverting the syringe. Discard the solution, and fill the syringe with de-ionized water up to the 2 mL graduation to continue the experiment until the next time point.
4. Determination of the vancomycin concentration
- Detect vancomycin concentration spectrophotometrically in deionized water at a peak absorption wavelength of 280 nm.
- Prepare known concentration range standards by adding 100 µL of 1 mg/mL stock solution to the UV 96-well plate and serially diluting using 100 µL of deionized water to obtain six levels of known concentrations (1 mg/mL to 0.03125 mg/mL).
- Transfer 100 µL of the eluents to UV-clear 96-well plates, and determine the concentration at 280 nm using a microplate reader.
- Generate a calibration curve by plotting the known drug concentrations against the corresponding absorbance values. Calculate the unknown drug concentration in the eluent using the linear equation generated by the calibration curve.
- Determine the drug mass by multiplying the concentration with the eluent volume (~1.7 mL).
- Calculate the cumulative drug release (mg) at a time point by summing the drug release from all the preceding time points.
- Normalize the drug release to the surface area of the strip (3.5 cm2) to determine the cumulative drug release per centimeter squared (cm2) of the implant.
5. Determination of the gentamicin concentration by the OPA tagging method 27
- Prepare GS standard solutions to perform the OPA assay.
- Transfer 1 mL of the 80 µg/mL GS solution to a centrifuge tube.
- Add 500 µL of PBS to five more centrifuge tubes.
- Perform a serial dilution with these tubes to obtain GS concentrations of 80 µg/mL, 40 µg/mL, 20 µg/mL, 10 µg/mL, 5 µg/mL, and 2.5 µg/mL. These serve as the calibration solutions for the assay.
- In a clear 96-well plate, add 80 µL of PBS to well A-1 and 80 µL of the calibration solutions to wells A-2 to A-7 in ascending concentration. This serves as an internal calibration for the assay.
- Add 80 µL of each sample to the empty wells in triplicate to be analyzed in the same 96-well plate. Limit the number of samples per well plate to <50 such that the timing of adding the OPA solution to the samples is approximately the same for all samples and to avoid evaporation.
- Add OPA reagent to all the standards and sample solutions.
- In a fume hood, fill a 25 mL reagent reservoir with methanol.
- Add 8 mL of methanol and 1 mL of the prepared OPA solution to a second reagent reservoir (section 1.8). Mix the solution thoroughly by pipetting up and down.
- With a multichannel 100 µL micropipette, add 48 µL of methanol from the first reservoir to all the sample-containing wells in the 96-well plate.
- Further to this step, add 72 µL of diluted OPA solution from the second reservoir to all the sample-containing wells in the 96-well plate. Incubate the well plate for 10 min at room temperature.
- Measure the fluorescence intensity (excitation of 340 nm, emission of 455 nm) using a microplate reader immediately after the 10 min incubation.
- Plot the calibration curve using the intensity readings for the solutions with known concentrations of GS. Add a linear line to determine the best fit and generate the corresponding equation.
- Calculate the drug concentration in the eluent from the calibration curves generated by plotting the known drug concentrations against the corresponding absorbance values.
- Calculate the drug mass by multiplying the concentration with the eluent volume (~1.7 mL).
- Determine the cumulative drug release (mg) at a time point by summing the drug release from all the preceding time points.
- Normalize the drug release to the surface area of the strip (3.5 cm2) to determine the cumulative drug release per centimeter squared (cm2) of the implant.
6. Bacteria preparation
NOTE: The following S. aureus strains were used in this study: type strain 12600, clinical strains L1101 and L1163 (obtained from Dr. Kerry LaPlante at the University of Rhode Island). The susceptibility profiles of these strains are presented in Table 1.
| Bacterial strains | Gentamicin MIC | Vancomycin MIC | |
| ATCC 12600 | ≤1 µg/mL (Sensitive) | ≤0.5 µg/mL (Sensitive) | |
| L1101 (Clinical strain) | ≥16 µg/mL (Resistant) | 8 µg/mL (Intermediate) | |
| L1163 (Clinical strain) | ≤1 µg/mL (Sensitive) | 8 µg/mL (Intermediate) | |
Table 1: Antibiotic susceptibility profiles of control and clinical S. aureus strains.
CAUTION: All the steps involving handling bacterial cultures and suspensions were performed in a BSL-2 lab space within a Class II, Type A2 Biosafety Cabinet.
- Thaw the bacterial stocks from −80 °C onto TSA plates to facilitate the growth of S. aureus. Statically incubate the plates overnight at 35 °C.
- Transfer two to three colonies from the overnight-grown agar plate cultures into 1 mL of sterile TSB, and statically incubate overnight at 35 °C.
- Transfer 100 µL of bacteria to a clear 96-well plate to spectrophotometrically measure the turbidity by determining the absorbance at 600 nm.
- Dilute the bacteria 10,000x using sterile MHB before determining the viable bacteria count using the luminescent assay.
7. Performing the real-time microbial viability assay
- Perform the luminescence-based assay using white opaque-bottom 96-well plates.
- Mix 100 µL of diluted bacterial suspensions with 100 µL of assay reagent prepared in section 1.5.
- Place a lid covered with aluminum foil over the prepared 96-well plate, and incubate for 5 mins with 100 rpm shaking.
- Measure the luminescence immediately with a microplate reader using the following settings in Gen5 3.11 software.
- Click 신규 to set up a protocol in the Task Manager window.
- Select Costar 96 white opaque in the drop-down menu for Plate Type.
- Click on Read under the Select Steps > Actions menu.
- Click on Luminescence within the Read Method window. Keep the Endpoint/kinetic and Luminescence fiber options selected under read type and optics type, respectively. Click on OK.
- Keep the <default> settings in the Read Step window (gain = 135; integration time = 1 sec; read height = 4.5 mm). Click on OK.
- The plate layout window appears ready for the run. Check the Use Lid box in the load plate window, and click on OK.
- Place the 96-well plate into the machine to read the luminescence values.
8. Generating a luminescence versus viable count standard curve for each bacterial strain
NOTE: Culture and enumerate all the S. aureus strains according to the methodology described in section 6.
- Prepare serially diluted bacterial suspensions to serve as standards.
- Enumerate the overnight-grown bacterial suspension by spectrophotometrically measuring the turbidity.
- Centrifuge at 6,000 × g for 5 mins to pellet the bacteria, and resuspend the pellet in sterile MHB to adjust the concentration to 1 x 109 CFU/mL.
- Dilute 100 µL of neat suspension using 900 µL of MHB, and perform a 10-fold serial dilution to reach 1 x 101 CFU/mL.
- Perform the luminescence assay on the prepared bacterial suspensions as described in section 7.
- Use sterile MHB as a blank control for the experiment, and subtract the blank luminescence value from the test sample luminescence values.
- Plot the log (CFU) against the log (luminescence) to generate a standard curve. Record the equation and R2 value.
- Determine the CFU/mL corresponding to the luminescence values using the strain-specific equation throughout the study.
9. Time-dependent antibacterial activity study setup
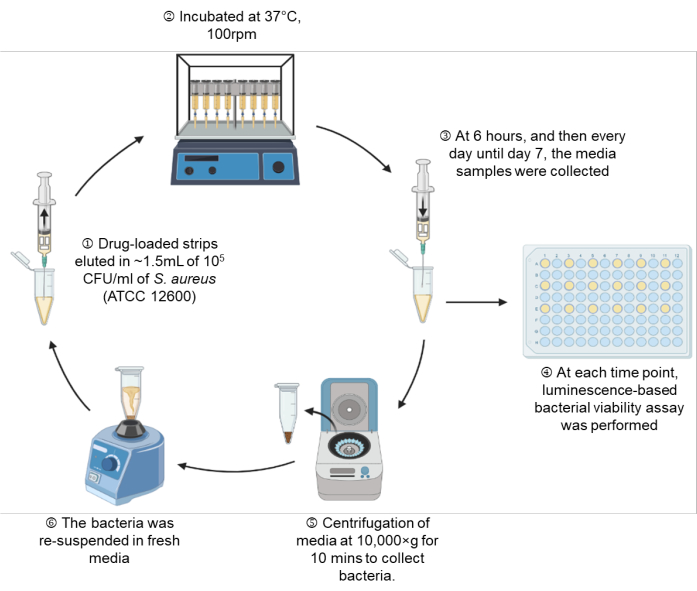
Figure 1: A schematic representation of the experimental setup. Please click here to view a larger version of this figure.
- Culture the bacteria overnight in 1 mL of TSB broth.
- Dilute the overnight grown bacterial suspension to 105 CFU/mL in sterile MHB (section 6), and verify the viable bacterial count using the luminescence units before the start of the experiment (section 7).
- Place the virgin UHMWPE and drug-loaded UHMWPE strips (3 mm x 5 mm x 10 mm, half the dimension of strips prepared for the elution studies in section 2) within a 3 mL syringe.
- Draw MHB containing 105 CFU/mL into the syringe through the attached needle up to the 1.5 mL mark, which is equivalent to 1.35 mL of MHB (Figure 1: Step 1).
- Place the syringe setup on a shaking incubator (100 rpm) at 37 °C until the indicated time points of 6 h, Day 1, Day 2, Day 3, Day 4, Day 5, Day 6, and Day 7 (Figure 1: Step 2).
- At each indicated time point, take out the syringe setup, and perform a real-time microbial viability assay according to section 7 on 100 µL of bacterial suspension (Figure 1: Step 3-4).
- Determine the viable bacteria count for the 6 h, Day 1, Day 2, Day 3, and Day 7 time points. Determine the CFU/mL from the luminescence units using the corresponding standard curve.
- Verify the absence of viable bacteria in the samples that showed luminescence values below the limit of detection by performing the spread-plate method on TSA plates and incubating at 35 °C overnight. Check for the presence of colonies the next day.
- Centrifuge the remaining bacterial suspension at 10,000 × g for 10 min, and gently aspirate the spent media. For tubes in which pellets are not visually observed due to the antibacterial activity of the drugs, leave 100 µL in the tube to ensure the unseen pellet is not disturbed (Figure 1: Step 5).
- Resuspend the pelleted bacteria in fresh MHB, and draw back the bacterial suspension into the same syringe setup through the attached needle (Figure 1: Step 6).
10. Quantification of adherent bacteria on the UHMWPE surface
- Retrieve virgin UHMWPE and drug-loaded UHMWPE surfaces from the syringe setup after the completion of the study on Day 7.
- Transfer the surfaces to 1.5 mL tubes, and rinse with 1 mL of sterile PBS (three times).
- Sonicate the surface for 40 min in 1 mL of sterile PBS. Determine adherent bacteria viability by performing a luminescence assay on 100 µL of the sonicated sample, as describedin section 7.
Following the protocol, UHMWPE was molded with gentamicin and vancomycin at 7% w/w and eluted into deionized water. The drug concentration in the eluent from the material was determined at 6 h, 1 day, 2 days, 3 days, and 1 week. The drug release from vancomycin and gentamicin-loaded UHMWPE demonstrated a burst release at 6 h followed by a steady release rate with a release concentration greater than the minimum inhibitory concentration (MIC) until 7 days (Figure 2, Table 2).
Prior to the antibacterial study, a standard curve was generated to correlate the luminescence units to the CFU/mL of the bacteria for each S. aureus strain (ATCC 12600, L1101, and L1163). The corresponding log (luminescence) values were plotted against the log (CFU/mL) to generate a standard curve (Figure 3). The R2 values calculated were 0.997, 0.996, and 0.994 for ATCC 12600, L1101, and L1163, respectively, indicating a strong fit for the concentration range. The equation derived was subsequently used to calculate the bacterial viability across all the experiments. ATCC 12600 and L1101 demonstrated a limit of detection within a range of 1 x 102-1 x 103 CFU/mL. On the other hand, the limit of detection for the L1163 strain was shown to be 1 x 104 CFU/mL.
The time-dependent antibacterial activity assay was performed using 1 x 105 CFU/mL as the starting inoculum for 12600, L1163, and L1101, which were exposed to 7% w/w drug-eluting materials for a period of 1 week. At each time point (6 h, 1 day, 2 days, 3 days, 1 week), the medium was refreshed, and the bacterial population was re-suspended. The exposure of the bacteria to the subsequent release of drugs from the material was continued until the next time point. UHMWPE with 7% w/w vancomycin and UHMWPE with 7% w/w gentamycin demonstrated >3log reduction for susceptible ATCC 12600 starting at 6 h, and complete eradication (no colony growth) was observed at the end of 3 days (Figure 4A). For the gentamicin-susceptible and vancomycin-intermediate strain L1163, both drug-eluting materials caused >3log reduction at 6 h, and complete eradication (no colony growth) was observed on day 1 of the experiment (Figure 4B). For the gentamicin-resistant and vancomycin-intermediate strain L1101, gentamicin elution from UHMWPE did not affect the bacterial viability of L1101 (Figure 4C). The bacteria proliferated, and the population stabilized within 6 h in the presence of virgin UHMWPE without antibiotic elution. In the presence of gentamicin-eluting UHMWPE, the population reached a similar growth level on day 2. On the contrary, vancomycin elution from UHMWPE significantly reduced the bacterial viability (>3log) at 6 h, and complete viability loss (no colony growth) was demonstrated by day 4.
The surfaces of both gentamicin-eluting and vancomycin-eluting UHMWPE showed no viable adherent bacteria when exposed to susceptible and intermediate-resistant strains after day 7 or complete eradication, whichever came first. Some viable bacteria (1 x 105 CFU/mL) were present on gentamicin-eluting UHMWPE exposed to gentamicin-resistant L1101. Similarly, approximately 1 x 105 CFU/mL of viable adherent bacteria were recovered from the control virgin PE (Figure 5).
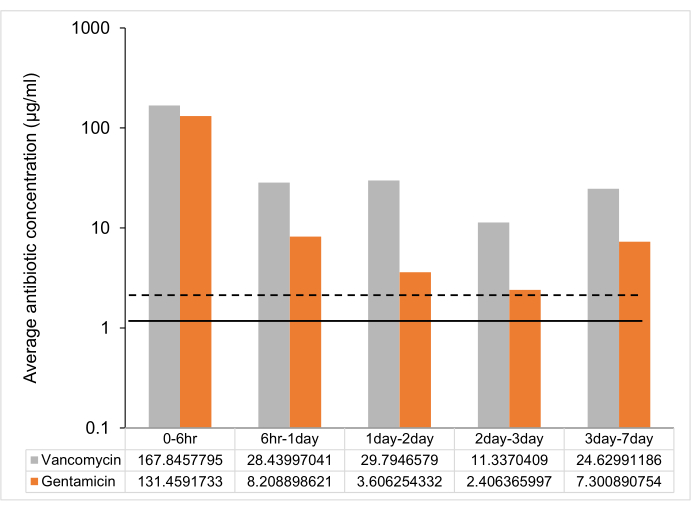
Figure 2: Time-dependent average antibiotic release from 7% w/w antibiotic-loaded UHMWPE strip. The average antibiotic release between time points from one 7% w/w gentamicin and vancomycin-loaded UHMWPE strip (3 mm3 x 5 mm3 x 10 mm3 ~ 2 cm2 surface area). The MIC against control strain ATCC 12600 is shown as a dotted line for gentamicin and a solid line for vancomycin. The error bars represent the standard deviation of the mean from six replicates (n = 6). Please click here to view a larger version of this figure.
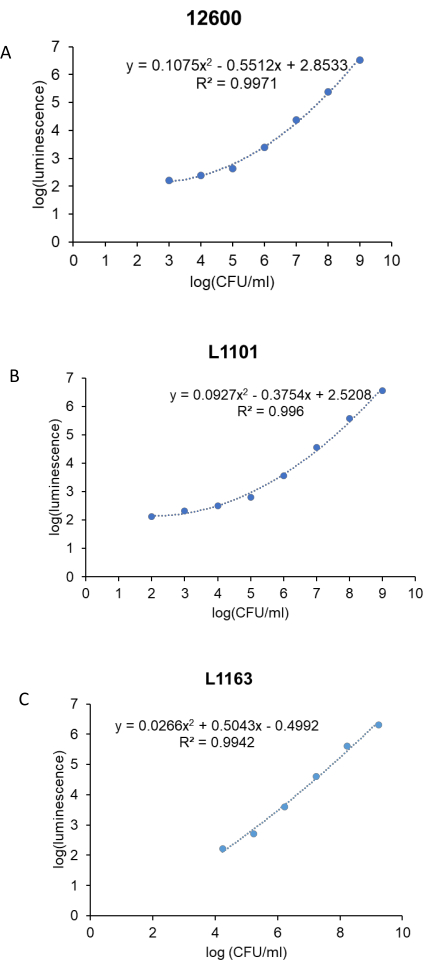
Figure 3: Real-time luminescent assay standard curve for all the S. aureus strains. Log (luminescence) was plotted against log (CFU/mL) to generate standard curves for control, (A) ATCC 12600, and clinical strains, (B) L1101 and (C) L1163. The equations describing the line of best fit and corresponding R2 values are indicated on the plots. Please click here to view a larger version of this figure.
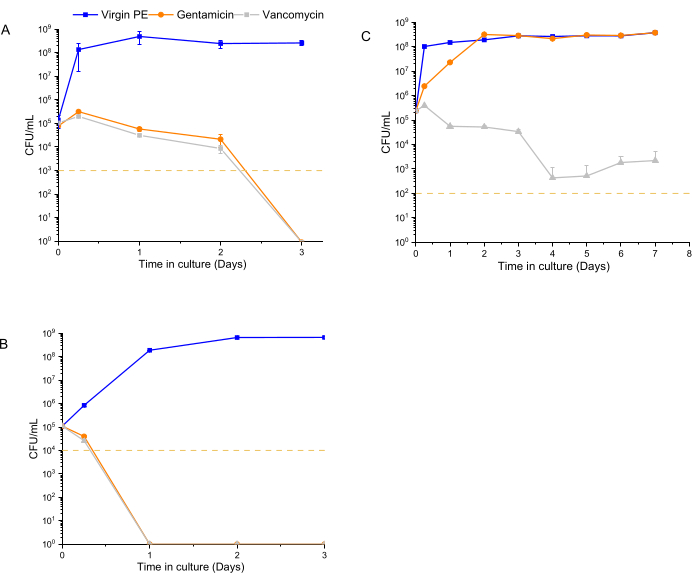
Figure 4: Bacterial viability determined using a luminescent assay for 7% w/w gentamicin-eluting and 7% w/w vancomycin-eluting UHMWPE. The time-dependent antibacterial activity of gentamicin and vancomycin eluted from UHMWPE against control strains, (A) ATCC 12600, and clinical strains, (B) L1163 and (C) L1101, are shown. Virgin 1020 PE served as a control for the experiment. The yellow line in the plots indicates the limit of detection for the respective S. aureus strain. Data are shown as mean ± standard deviation (n = 3). Please click here to view a larger version of this figure.
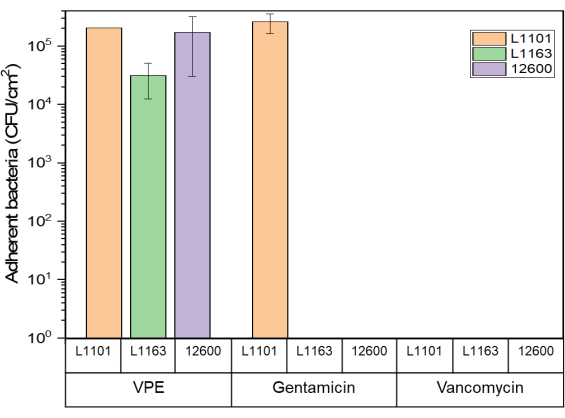
Figure 5: Adherent bacteria viability determined using the luminescent assay Glo assay for 7% w/w gentamicin-eluting and 7% w/w vancomycin-eluting UHMWPE against all S. aureus strains. The bar chart indicates adherent bacteria (CFU) recovered per centimeter squared (cm2) of 7% gentamicin-loaded and 7% vancomycin-loaded UHMWPE at the end of the study period for all the strains tested. The bars show data as mean ± standard deviation (n = 3). Please click here to view a larger version of this figure.
| Time points | Vancomycin | Gentamicin |
| Peak concentration (µg/mL) | Peak concentration (µg/mL) | |
| 0 – 6 h | 336 ± 72 | 263 ± 24 |
| 6 h -1 day | 57 ± 18 | 16 ± 2 |
| 1 day – 2 day | 60 ± 18 | 7 ± 1 |
| 2 day – 3 day | 23 ± 6 | 5 ± 0.4 |
| 3 day – 7 day | 49 ± 20 | 15 ± 1 |
Table 2: Peak drug concentration (μg/mL) at different time points. Data are shown as mean ± standard deviation (n = 6).
Supplementary Figure 2: Luminescence signal decay over a period of 10 min from the addition of assay reagent to the sample. A ±5% difference in the signal is shown as a dotted line Please click here to download this File.
