1. Preparation of Materials
The material necessary for vampiric reverse microinjection is similar to that required for standard microinjection techniques used to make transgenic C. elegans strains 1. Although some reagents (e.g. assay plates) are made the day of experimental transfer, many of the materials must be coordinately prepared over an 8 day period (see Table 1 for a time table). As such, it is important to plan ahead carefully when using this technique (for necessary reagents and equipment see Table 2).
Injection pads:
- Make a 2% agarose solution in H2O and heat until dissolved. Aliquot in 1ml aliquots in 1.5 mL microcentrifuge tubes and store at 4 °C.
- Lay out 22 x 50 mm cover glasses on a bench top with their edges slightly protruding off the bench top edge to facilitate picking them up quickly.
- Poke a small hole in the lid of the microcentrifuge tube (use a thumbtack) to allow venting. Place the tube in a 15 mL beaker with approximately 5 mL of water, and microwave to melt (approximately 35 seconds).
- Using a Pasteur pipette and bulb place a drop (approximately 35 μL) of the melted agarose on a cover glass and immediately place a second cover glass over the drop at a 90° angle to the first. Repeat for several other cover glasses.
- After the agarose has solidified, remove the cover glass and allow the pad to air dry completely overnight (if needed sooner, slides can be placed in a 50-80 °C oven for 15-30 minutes).
- Pads can then be stored in a cover glass box at room temperature indefinitely.
Assay plates:
For assaying the embryonic lethality associated with pal-1 RNAi, the preparation of assay plates should be done on the day of the vampiric experiment. The goal is to have a minimal bacterial lawn while not starving your worms. An overly thick lawn makes scoring Pal-1 larvae extremely difficult as the small, translucent deformed L1 animals can easily be lost in the food. Plate preparation should be optimized for scoring the phenotype of interest.
- Prepare OP50: Seed 5mLs of LB broth with a single OP50 colony grown on LB agar plates. Incubate the OP50 at 37 °C overnight while shaking, then store at 4 °C.
- Prepare 35 mm NGM plates according to basic protocol (see Note 1).
- Spot 20 μL of the overnight OP50 culture (step 1.7) onto each NGM plate. Allow the LB-OP50 to dry (this should take less than 20 minutes).
Injection Needles:
- Pull injection needles from borosilicate glass capillaries using a P97 flaming/brown micropipette puller from Sutter instruments (see Figure 1 for representative needle shape).
- Store injection needles in a needle holder constructed from a Petri plate and modeling clay (see 1).
2. Preparation of Worms
The estimated pseudocoelomic volume of an adult C. elegans hermaphrodite is 40-80 picoliters (pers com. David Hall). To obtain samples of extracellular fluid from such a small reservoir it is beneficial to increase the total available resource. We have identified three methods to greatly increase the available fluid in the donor worms. Our primary method exploits the phenotype of clr-1(e1745) mutants, which can cause a 10-fold or greater increase in extracellular fluid volume (Figure 2). The two alternative methods utilize the fact that the germline accounts for almost one third of the total volume of the worm, and its removal by glp-1(RNAi) or laser ablation results in animals with pseudocoelomic fluid filling the germline void (Figure 2) 2.
Preparation of donor worms using clr-1(e1745)
See Figure 3 for clr-1(e1745) transfer assay workflow
- Streak out bacteria expressing dsRNA for single colonies on LB + 25μg/mL carbenicillin plates. We will use pal-1 dsRNA producing bacteria for this demonstration. Incubate overnight at 37 °C.
- From a streaked LB+ 25 μg/mL carbenicillin plate of pal-1(RNAi) bacteria, pick a single colony to inoculate a 3 mL overnight culture in LB supplemented with 25μg/mL carbenicillin.
- Pipette 15 μL of overnight culture onto 35mm NGM plates supplemented with 25 μg/mL carbenicillin and 1 mM IPTG. Make sure to plate the bacteria off center of the plate. Allow to dry (approximately 20 minutes).
- Make 500 μL fresh bleach solution (1:1 5M KOH: NaHypochlorite).
- Pipette 10 μL of bleach solution on the seeded plate. Be sure to place the bleach droplet away from the bacterial spot.
- To obtain a small, clean, synchronized population of clr-1(e1745) animals for reverse microinjection, pick 2-10 gravid adults into the bleach drop. The adults dissolve, leaving clean, partially developmentally staged embryos. These embryos will then hatch and the larvae will crawl to the food.
- Incubate for four days at 20 °C.
- Shift plates to 25 °C for four hours to induce swelling.
- Pick worms to an unseeded NGM plate and allow time to clear off bacteria from the cuticle. (OPTIONAL STEP) Rinse worms in M9 while transferring to NGM plates.
- Move L4/Young Adult (YA) recipient worms to an unseeded plate NGM plate and allow time to clear off bacteria from the cuticle. (OPTIONAL STEP) Rinse worms in M9 while transferring to NGM plates.
A2) Alternative preparation of donor worms using glp-1(RNAi)
- From a streaked plate of LB + 25 μg/mL carbenicillin inoculate a 3 mL overnight culture of LB supplemented with carbenicillin with a single colony of glp-1(RNAi) bacteria. Incubate at 37 °C overnight.
- Pipette 15 μL of glp-1(RNAi) overnight culture onto a 35 mm NGM plate supplemented with 25 μg/mL carbenicillin and 1mM IPTG. Incubate at room temperature overnight.
- Transfer five L3/L4 animals to the glp-1(RNAi) plates. Incubate for two days at 20 °C. The RNAi targeting glp-1 results in progeny with germline proliferation defects. The inability to fully develop the germline results in unoccupied space that fills with extracellular fluid. Optimization of the RNAi food to generate progeny that progress to adulthood without proliferative germlines may be necessary depending.
- Have prepared pal-l(RNAi) plates as in steps 2.1-2.3 of the clr-1(e1745) worm preparation protocol above.
- Make a fresh bleach solution (1:1 5M KOH: NaHypochlorite).
- Pipette 10 μL of bleach solution on the seeded pal-1(RNAi) plate. Be sure to place the bleach droplet away from the bacterial spot.
- Transfer embryos by picking 2-10 gravid glp-1(RNAi) adults into the bleach droplet.
- Incubate for four days at 20 °C.
- Pick worms to an unseeded 60mm NGM plate and allow time to clear off bacteria from the cuticle. (OPTIONAL STEP) Rinse worms in M9 while transferring to NGM plates.
B2) Alternative preparation of donor worms using germline laser ablation
- Streak out bacteria expressing dsRNA for single colonies on LB + 25 μg/mL carbenicillin plates. We will use pal-1 dsRNA producing bacteria for this demonstration. Incubate overnight at 37 °C.
- From a plate with carbenicillin inoculate a 3 mL overnight culture in LB supplemented with 25 μg/mL carbenicillin.
- Pipette 15 μL of the overnight culture of pal-1 dsRNA-expressing bacteria onto 35 mm NGM plates supplemented with 25 μg/mL carbenicillin and 1mM IPTG. Allow to dry overnight.
- Isolate L1 animals from standard OP50 plates and laser ablate the somatic germline precursor cells Z1 and Z4 using a standard protocol 3.
- Recover laser ablated worms on the aforementioned pal-1 RNAi plates.
- Incubate for 3 days at 20 °C.
- Pick worms to a clean NGM plate and allow time to clear off bacteria from the cuticle. (OPTIONAL STEP) Rinse worms in M9 while transferring to NGM plates.
Preparation of recipient worms
- Streak out OP50 for single colonies on LB plates. Incubate overnight at 37 °C.
- From the LB plate inoculate a 3 mL overnight culture in LB.
- Pipette 15 μL of the overnight culture onto 35 mm NGM plates. Make sure to plate the bacteria off center of the plate. Allow to dry (approximately 20 minutes).
- Make a fresh bleach solution (1:1 5M KOH: NaHypochlorite).
- Pipette 10 μL of bleach solution on the seeded plate. Be sure to place the bleach droplet away from the bacterial spot.
- Transfer embryos by picking 2-10 gravid N2 adults into the bleach droplet.
- Incubate for three days at 20 °C.
- Move worms to a clean, unseeded NGM plate and incubate at 20 °C for at least 30 minutes.
3. Vampiric Isolation of Extracellular Fluid
The following protocol is specific to a microinjection set-up consisting of a pli-100 pico-injector, a stationary needle held in a micromanipulator, and a floating stage in which the worm can be slid into the needle. However, the generalized technique is to insert an empty microinjection needle into a donor animal, while maintaining sufficient pressure to prevent capillary in flow of mineral oil prior to entry and cellular material while penetrating the body-wall tissue. While the needle is within a donor animal pressure is reduced to allow capillary filling of the needle with extracellular fluid, which can then be moved in the needle to a recipient worm and expelled by increasing the pressure sufficiently. Removal of the fluid by capillary action can also be assisted by using the fill, or suction, function of the pico-injector. This generalized technique should be easily adaptable to other micro-injection systems that allow control of balance pressure.
- Turn on the inverted microscope, the dissection microscope, and pico-injector (See Figure 1).
- Turn the selector knob of the Pico injector to Pclear, and check that the current measurement is in psi. This reading, the clear pressure, is the input pressure and should be approximately 0 psi.
- Check the primary valve on the nitrogen tank regulator to make sure that the output is closed (turn counterclockwise until knob is loose).
- Open the main nitrogen tank valve. The regulator gauge nearest the N2 tank should now read the internal pressure of the tank.
- Slowly increase the out pressure by turning the primary regulator valve clockwise. Monitor the increasing pressure on the pico-injector (this is more accurate than the output pressure reading on the regulator gauge). Slowly increase the pressure to 100 psi. DO NOT ALLOW TO EXCEED 105 psi.
- When 100 psi is reached switch the selector to Pinject and set the injection pressure to 30 psi using the Pinject knob.
- Switch the selector knob to Pbalance.
- Place a 15 μL droplet of mineral oil on your clean donor and recipient plates.
- Cover your 2% agarose injection pad in mineral oil.
- Load your pulled micropipette needle in the holder and align.
- Using a dissection microscope, mount 1 donor and 1 recipient worm near each other on the agarose pad.
- Position worms using low magnification and bring needle into the mineral oil near the donor.
- Move to high power and position needle near a pseudocoelomic cavity.
- Increase the balance pressure to approximately 10 psi. Note a mineral oil plug at the tip of the needle.
- Push the worm into the needle to position the needle tip in the pseudocoelomic cavity by moving the stage.
- Note a jump in the position of the mineral oil in the tip of the needle.
- Reduce the balance pressure to allow capillary action to fill the needle. (One can also use the fill function to speed the process).
- After collecting fluid slide the worm away from the needle.
- Switch to low power magnification and position the needle by the recipient worm (DO NOT ALLOW THE NEEDLE TIP TO LEAVE THE MINERAL OIL). [Alternatively, the needle holder can be removed and the fluid transferred to a droplet in a microcentrifuge tube.]
- Switch back to high magnification and move the needle into the recipient worm by sliding the microscope stage.
- Once positioned in the recipient worm use the injection (set at 35 psi) function to inject the pseudocoelomic fluid collected from the donor.
- Remove the needle from the recipient worm by sliding the microscope stage in the opposite direction as used to enter the recipient.
- With the needle out of the worm, raise the needle away from the injection pad.
- Remove the injection pad and place a droplet of M9 on the worms to recover.
- Place a 10 μL droplet of M9 on an assay plate.
- Pick the recipient worm into the M9 on the assay plate.
4. Assaying RNAi Phenotypic Transfer
- Allow recovered worms to grow for 24 hours at 20 °C.
- Remove recipient adults, leaving embryos and hatched progeny on the plate. Incubate plate for an additional 24 hours at 20 °C.
- Score progeny as having not hatched, hatched but being phenotypically mutant, or being wild-type larvae.
Notes
- NGM media is made in 1-liter batches by adding 18 g of agar, 2.5 g of bactopeptone, 3 g of sodium chloride and H2O. Add stir bar and autoclave. After autoclaving put the flask on a stir plate and allow the media to cool to 60 °C while stirring. After the media has cooled, add 1 ml of cholesterol (5 mg/ml in ethanol), 1 mL of 1M CaCl2, 1 mL of 1M MgSO4, 25 mL of potassium phosphate buffer (pH6.0). If plates are to be used for RNAi the NGM is supplemented with 1 mL of 1M IPTG and 1 mL of 25 mg/mL carbenicillin after cooling to 60 °C. Plates are poured in either 35 mm (3.5 mL of NGM) or 60 mm (8.5 mL of NGM) Petri plates.
- 2% agarose made in water. Store in fridge. Melt and make pads well in advance of the day of pseudocoelomic fluid isolation. The dryness of the pad makes the worm adhere to the pad, and a minimum of one day of air drying is necessary for the pads to be dry enough. If the injections pads are too dry and your donor worms are desiccating faster than you can manipulate them you can add additional moisture to injection pads by breathing on the pad before adding mineral oil. Regardless the ability to work quickly to isolate pseudocoelomic fluid before the worm dries is absolutely necessary.
- We use carbenicillin at all steps of our RNAi food preparation to select for the ampicillin resistance marker. Carbenicillin is an ampicillin analog which is more stable and results in fewer satellite colonies than ampicillin.
- We use RNAi plates which are NGM supplemented with 1mM IPTG and 25 μg/mL carbenicillin. The RNAi feeding vectors are in the HT115 E. coli strain which is deficient for a dsRNA specific nuclease.
- (Optional Step) We find that given an hour on unseeded NGM plates donor and recipient worms do an adequate job losing cuticle bound bacteria that is carried over from the feeding plates. This is not true though in cases where the RNAi target in the donor animal interferes with mobility (e.g. unc-22(RNAi)). It therefore becomes necessary to assist in the process. To do so, we use a depression slide with approximately 50 μL of M9. Picking worms into the M9 from the RNAi feeding plates, then stirring with a standard platinum worm pick is sufficient to remove most adherent bacteria.
5. Representative Results
Presented are representative results for experimental transfer for extracellular fluid from clr-1(e1745) worms grown on bacteria expressing dsRNA targeting pal-1. The progeny of the donor animals died and/or exhibited posterior patterning defects. We then transferred extracellular fluid from these animals to the pseudocoelom of RNAi naïve wild-type worms. A portion of the subsequent progeny of the recipient animal then displayed the expected pal-1 mutant phenotypes (Figure 4). This is in contrast to the progeny of recipient animals who received extracellular fluid from donor animals grown on either standard bacterial food or control RNAi vector bacteria who displayed only background levels of lethality (Figure 4). While progeny of recipients of extracellular fluid from donor animals undergoing RNAi show a significant increase in the frequency of dsRNA induced phenotypes, the penetrance is not as strong as in the donor animals’ progeny, where nearly 100% of progeny die as unhatched embryos, and only rare, severely deformed animals hatch.
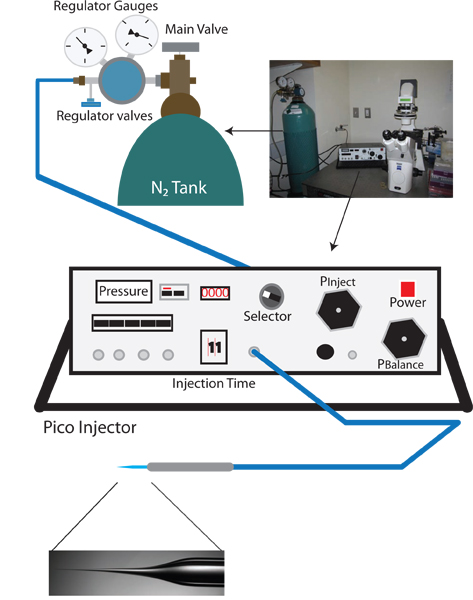
Figure 1. Vampiric reverse injection setup. The appropriate setup for a vampiric reverse microinjection setup is similar to a standard C. elegans microinjection rig, and includes a dissection microscope, an inverted microscope with 10X and 40X objectives, a moveable stage for positioning, and a micromanipulator with injection needle holder. Additionally, a needle puller for preparation of injection needles is also needed. Unique to the reverse microinjection protocol a pico-injector which allows fine control of balance pressure (e.g. Warner Instruments PLI-100) is needed.
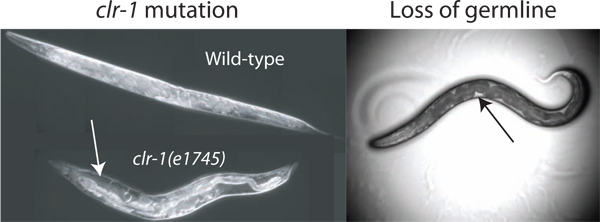
Figure 2. Enhancement of pseudocoelomic volume. The volume of pseudocoelomic fluid available for isolation is insufficient for assay in wild-type animals. The volume can be increased by disrupting the regulation of osmotic balance through temperature shift of clr-1(e1745) animals, resulting in obvious accumulation of pseudocoelomic fluid (white arrow). Additionally the loss of the gonad by laser ablation or glp-1(RNAi) enables access to extracellular fluid (loss by laser ablation is shown). The available fluid is most readily observed as clear patches between the dark intestine and the body wall (black arrow), although the total available volume is much less than that found in clr-1(e1745) animals grown at the restrictive temperature.
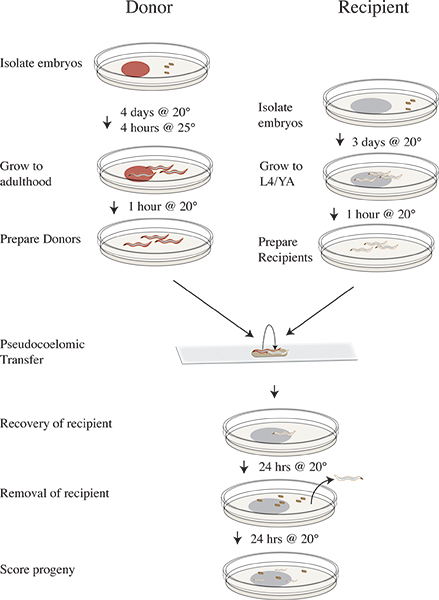
Figure 3. Vampiric isolation and transfer protocol timeline. Four days prior to transfer experiment isolate clr-1(e1745) donor embryos on plates with RNAi food and incubate at 20 °C. Three days prior isolate N2 recipient embryos on OP50. On the day of the experiment shift the donor plates to 25 °C for four hours. Remove the donors after 25 °C incubation to clean plates lacking food and shift to 20 °C. Move the recipient animals to clean plates lacking food and continue incubating at 20 °C. Perform transfer experiment and recover recipient worms on OP50 plates and incubate at 20 °C. Remove the recipient animals after 24 hours at 20 °C. Incubate recipient progeny for 24 hours at 20 °C. Score progeny as wild-type, mutant, or unhatched.
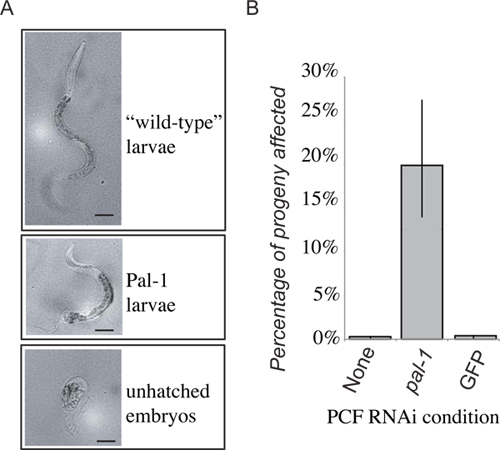
Figure 4. Representative results. The loss of PAL-1 function in the recipient will result in embryonic lethality, or distinctive loss of posterior development (A). 48 hours after PCF transfer progeny laid within the first 24 hours are scored as either wild-type larvae, Pal-1 larvae, or unhatched embryos. The frequency of unhatched embryos and phenotypically Pal-1 larvae are combined to give a measure of pal-1(RNAi) transfer induced phenotypes. Receipt of pseudocoelomic fluid from animals grown on PAL-1 dsRNA food produces a strong induction of associated phenotypes unseen in control transfer recipients (B).
