1. High-pressure Polytene Chromosome Preparation from Ovarian Nurse Cells
- Dissect half-gravid Anopheles females at 25 hours post-blood feeding under a dissection microscope and fix ovaries in fresh modified Carnoy’s solution with methanol (100% methanol: glacial acetic acid, 3:1). At this stage the ovaries are at Christophers’ III stage of development when follicles have an oval shape and a transparent area with nurse cells within follicles has a round shape13. Put ovaries from approximately five females into 500 μl of modified Carnoy’s solution in a 1.5 ml Eppendorf tube and keep them at room temperature for 24 hours. Transfer ovaries to -20 °C for a long-term storage.
- Prepare modified Carnoy’s solution with ethanol (100% ethanol: glacial acetic acid, 3:1) and 50% propionic acid just prior to making slides. Place one pair of ovaries on a dust-free slide in one drop of modified Carnoy’s solution. Split ovaries into approximately 6 sections with dissecting needles and place them into drops of 50% propionic acid on clean slides under a dissection MZ6 Leica stereomicroscope. Use a separate slide for each section.
- Separate follicles dissecting using needles and wipe out remaining tissue with filter paper or paper towel under a dissection microscope. Add a new drop of 50% propionic acid to the follicles and allow them to sit for 3-5 minutes at room temperature. Place a coverslip on top of the 50% propionic acid droplet. Let the slide stand for approximately 1 minute.
- Wrap the slide with filter paper and plastic. Using a Dremel 200 rotary tool with a Flex-Shaft attachment and soft plastic tip set between 3000-5000 RPM, express the follicles by swirling the tip in circles and lightly pressing the coverslip to evenly spread the nuclei. This step should take approximately 1 minute. Check the spread quality with an Olympus CX41 Phase Microscope using a 20x objective.
- Prepare a sandwich by placing an additional coverslip next to the coverslip covering the chromosomes, and cover them with a second microscope slide. This reduces the chance of crushing the slide in the vise. Wrap the slides with a plastic sheet that come in glass slide containers and filter paper to hold the sandwich and to protect the slide from scratching due to the vise.
- Apply pressure to the slides via mechanical vise. A pressure of 85-120 inch-lbs is sufficient and is achieved by using a torque wrench. This step is necessary for flattening the chromosomes as much as possible.
- Remove the second microscope slide and the additional coverslip. Heat the slide with the coverslip covering the chromosomes to 55 °C on a slide denaturation/hybridization system for 10-15 minutes to further flatten the chromosomes. Dip the slide in liquid nitrogen for at least 15 seconds, and, when bubbling has stopped, proceed to quickly remove the coverslip with a razor blade. Immediately place slides in cold 50% ethanol for 5 minutes. Dehydrate slides in 70%, 90%, 100% ethanol for 5 minutes each. Air dry slides.
2. FISH Using an Automated Slide Staining System
Program is set up with the Xmatrx automated slide staining system to run the following steps, except the probe labeling. The detailed nick-translation labeling protocol is described in the documentation provided with DNA polymerase from Fermentas.
- Before FISH, prepare fluorescent probes by labeling genomic BAC DNA with a fluorochrome using a nick-translation protocol. Mix 1 μg of DNA, 0.05 mM each of unlabeled dATP, dCTP, and dGTP and 0.015 mM dTTP, 1 μl Cy3- or Cy5-dUTP, 0.05 mg/ml BSA, 5 μl of 10x nick-translation buffer, 20 units of DNA polymerase I, 0.0012 units of DNase I, and nuclease-free water to 25 μl. The DNA polymerase I/DNase I ratio is selected empirically to obtain probes with a size range from 300 to 500 bp. Incubate the mix at 15 °C for 1 hour.
- Put slides and reagents into the Xmatrx automated slide staining system and start the program to run the following steps. Apply 800 μl of 1x PBS for 20 min. Blow slides with air. Perform formalin fixation by applying 450 μl of 4% formalin in 1x PBS for 1 min followed by washes with 100% ethanol for 1 sec twice and for 2 min once. Blow slides with air.
- Heat slides at 45 °C for 2 min to avoid bubbling when applying the probes. Apply 20 μl of the DNA probes, add drops of mineral oil to avoid evaporation of a hybridization solution, and place a coverslip on top. Denature chromosomes and DNA probes by heating slides at 90 °C for 10 min.
- For hybridization, incubate slides at 42 °C for 14 hours with coverslips on. The hybridization solution consists of 2x SSC, 100 mM sodium phosphate, 1x Denhardt’s solution, 100 μg/ml of sodium azide, and 10% dextran sulfate in formamide.
- For stringency washes, heat slides at 42 °C for 2 min, remove coverslips, wash slides in 2x SSC for 1 sec 4 times. Blow slides with air. Apply 800 μl of 0.4x SSC at 42 °C for 10 min 2 times. Wash slides in 2x SSC at 25 °C for 10 min.
- Perform chromosome staining by applying 50 μl of 1 μM YOYO-1 in 1x PBS. Apply drops of mineral oil to avoid evaporation of the staining solution, put on coverslips, and incubate at 25 °C for 10 min. Remove coverslips, wash slides in 2x SSC for 1 sec 4 times. Blow slides with air. Apply 15 μl of ProLong Gold antifade reagent. Put on coverslips.
3. Slide Reading with an Automatic Fluorescent Imaging System
This section details the use of the Duet software, which comes standard with the ACCORD PLUS automated scanning system. Instructions begin after turning on in this order: Olympus U-RFL-T power supply for a halogen bulb, computer Dell precision T3500, microscope Olympus BX61 with a connected camera Olympus U-CMAD3.
- For setting up 10x Pre-Scan, open the Duet software in the ACCORD PLUS automated scanning system. Click “Online” button. Enter new Case ID and assign a Slide ID. Click the dot labeled “BF.” This is the brightfield option. Set a scan choice to “10x Prescan.” Use “2500x circle,” “10000x circle” or “rectangle.” Click “Set&run” for 10x Pre-Scan.
- Click the “OK” button to run 10x Pre-Scan. Follow the prompts to adjust the scanning properly. Click “Finish” to start the scan. Press the “Main” tab to go back to the main screen. Click “Offline” button. Find the Case ID and Slide ID that was assigned and click “Offline Scan.” In the black box at the top left area click on an arrow and select “10x Prescan.” Using the arrows (< || Δ > a.k.a. “back,” “pause,” “play,” “forward” buttons), go through the scanned images.
- After finding an image of interest, double click on the screen the middle of the target region and press “Snap.” This will target an image for capture later on. After selecting all targets, click “Classify.” Select “10x Prescan.” Right click an image and get the chance to classify the images. Select “Polytene.”
- Set up 40x Pre-Scan in the Duet software. Click “Main” to go back to the main menu. Select “Online” again. Select a slide. Change “BF” to “FL.” Change the task name to “Revisit-X40-RG.” Change the section right below the last setting to “Revisit-ALL.” Click “Set&Run.” Press “OK.” Follow the prompts again to set up the automation. Click the “Start matching views” button to match 10x and 40x images. Click “Finish” to start the scan. Once done, click “Classify” and look at the images.
4. Representative Results
Figure 1 graphically depicts the scheme of the high-pressure chromosome preparation. This step involves the process of squashing and flattening chromosomes using a Dremel tool and mechanical vise, as well as chromosome visualization using a phase-contrast microscope. Figure 2 illustrates the hybridization of a fluorescently labeled probe to target DNA on the chromosome slide preparations using an automated slide staining system, the use of an automated scanning microscope for visualizing and mapping the probes after the FISH experiment, and placing and orienting genomic scaffolds on the chromosomes.
Preparations of ovarian nurse cell polytene chromosomes from females of An. gambiae were made using the high-pressure technique. This method does not damage or change most of the chromosome structure (Figure 3). It flattens bended chromosomes and, thus, reveals hidden fine bands that are not seen on regular preparations (Figure 4).
The probes used in this protocol are genomic BAC DNA clones obtained from a ND-TAM BAC library generated from An. gambiae PEST strain DNA. The genomic DNA for this library was extracted from newly hatched first instar larvae of both sexes. Figure 5 shows the results of FISH using BAC clones hybridized to polytene chromosomes of An. gambiae. This procedure was performed using the Xmatrx automated slide staining system. Using a cytogenetic photomap for An. gambiae10, two BAC clones, 102B24 (GenBank: BH372701, BH372694) and BAC 142O19 (GenBank: BH368703, BH368698), were localized in subdivisions 16C and 16D of the 2R arm, respectively. However, the BLAST search against the An. gambiae PEST strain AgamP3 assembly 14 identified the sequences homologous to 102B24 and 142O19 in subdivisions 16B and 17A of the 2R arm, respectively (Table 1). Therefore, the correspondence between the genomic coordinates and the cytogenetic subdivisions can now be adjusted according to our mapping data. The BLAST search against the An. gambiae M form and S form genome assemblies found that the two BAC clones are located in different contigs, but in the same scaffolds within each form (Table 1). The identified contigs can now be associated with specific chromosomal locations. Moreover, the identified scaffolds can now be properly oriented within the cytological subdivisions 16CD. Interestingly, the distances between the 102B24 and 142O19 sequences in the scaffolds are 1,892,981 bp, 1,658,391 bp, and 1,688,426 bp in the PEST strain, M-form, and S-form genome assemblies, respectively. Because, the PEST strain is a hybrid between the M and S forms, this difference is likely due to incorrect assembly of the PEST genome.
| BAC (accession) |
PEST-strain AgamP3 Coordinates |
M-form contigs Coordinates |
M-form scaffolds Coordinates |
S-form contigs Coordinates |
S-form scaffolds Coordinates |
| 102B24 (BH372694) |
2R:AAAB0100 8844_26 (16B) 43,681,342- 43,681,874 |
ABKP020247 53.1 32,513- 33,045 |
EQ090167.1 1,858,377- 1,858,909 |
ABKQ010123 32.1 4,299- 4,832 |
EQ099711.1 215,891- 216,424 |
| 102B24 (BH372701) |
2R:AAAB0100 8844_28 (16B) 43,788,213- 43,788,969 |
ABKP020247 51.1 24,816- 25,575 |
EQ090167.1, 1,772,683- 1,773,442 |
ABKQ010123 35.1 27,782- 28,540 |
EQ099711.1, 305,514- 306,272 |
| 142O19 (BH368698) |
2R:AAAB0100 8805_5 (17A) 45,428,580- 45,429,264 |
ABKP020247 01.1 2,499- 3,185 |
EQ090167.1 315,806- 316,492 |
ABKQ010123 76.1 49,242- 49,942 |
EQ099711.1 1,791,625- 1,792,325 |
| 142O19 (BH368703) |
2R:AAAB0100 8805_8 (17A) 45,560,099- 45,574,323 |
ABKP020220 17.1 30,971- 32,469 |
EQ090167.1 200,518- 202,016 |
ABKQ010123 79.1 27,760- 28,508 |
EQ099711.1 1,903,569- 1,904,317 |
Table 1. BLAST results of the BAC-end sequences against the genome assemblies of An. gambiae.

Figure 1. Schematic representation of high-pressure chromosome preparation. Mosquito ovaries are shown at the correct stage of development.
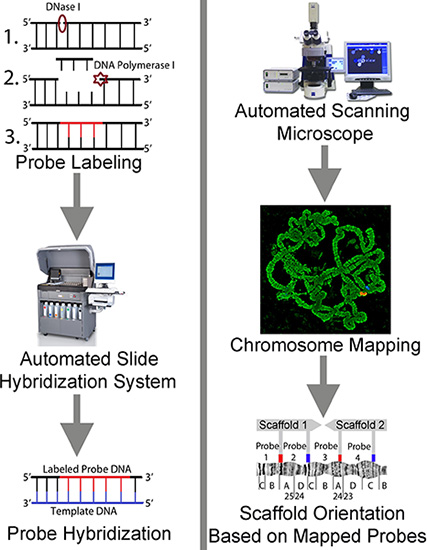
Figure 2. A scheme representing automated FISH, slide scanning, and chromosome mapping of genomic scaffolds.
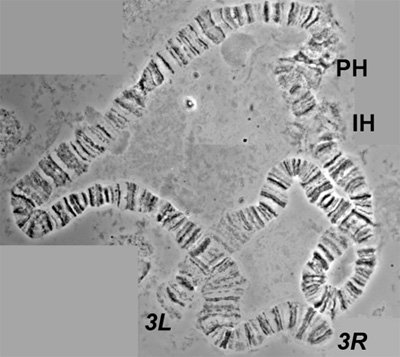
Figure 3. A spread of polytene chromosome 3 of An. gambiae obtained using the high-pressure technique. 3L and 3R mark left and right arms at their telomeres. PH and IH indicate pericentric and intercalary heterochromatin, respectively.
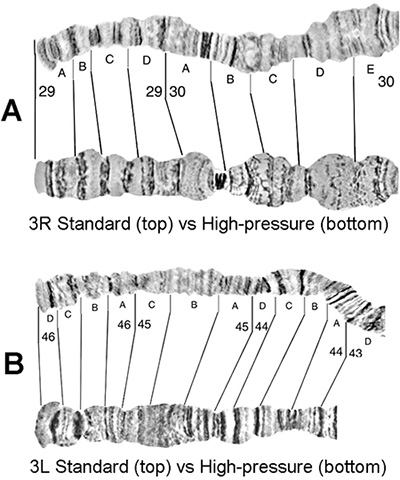
Figure 4. Comparison of An. gambiae polytene chromosomes prepared using the traditional (top) and high-pressure (bottom) technique. The images cover subdivisions 29A-30E of arm 3R (A) and 43D-46D of arm 3L (B).
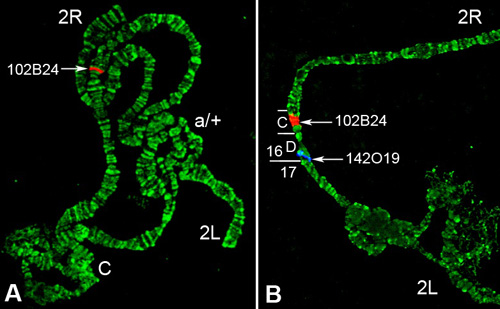
Figure 5. FISH of BAC clones to polytene chromosomes of An. gambiae. A) Hybridization of 102B24 (red signal) with the 2R arm. B) Dual-color FISH of 102B24 (red signal) and 142O19 (blue signal) to subdivisions 16C and 16D of the stretched 2R arm, respectively. Arrows indicate signals of hybridization of BAC clones labeled with Cy3 (red) and Cy5 (blue). C-the centromeric region. a/+ shows the heterozygote 2La inversion. Chromosomes were counterstained with the fluorophore YOYO-1.
