Infinium Assay for Large-scale SNP Genotyping Applications
Summary
A protocol is described that uses Illumina's Infinium assays to perform large-scale genotyping. These assays can reliably genotype millions of SNPs across hundreds of individual DNA samples in three days. Once generated, these genotypes can be used to check for associations with a variety of different diseases or phenotypes.
Abstract
Genotyping variants in the human genome has proven to be an efficient method to identify genetic associations with phenotypes. The distribution of variants within families or populations can facilitate identification of the genetic factors of disease. Illumina's panel of genotyping BeadChips allows investigators to genotype thousands or millions of single nucleotide polymorphisms (SNPs) or to analyze other genomic variants, such as copy number, across a large number of DNA samples. These SNPs can be spread throughout the genome or targeted in specific regions in order to maximize potential discovery. The Infinium assay has been optimized to yield high-quality, accurate results quickly. With proper setup, a single technician can process from a few hundred to over a thousand DNA samples per week, depending on the type of array. This assay guides users through every step, starting with genomic DNA and ending with the scanning of the array. Using propriety reagents, samples are amplified, fragmented, precipitated, resuspended, hybridized to the chip, extended by a single base, stained, and scanned on either an iScan or Hi Scan high-resolution optical imaging system. One overnight step is required to amplify the DNA. The DNA is denatured and isothermally amplified by whole-genome amplification; therefore, no PCR is required. Samples are hybridized to the arrays during a second overnight step. By the third day, the samples are ready to be scanned and analyzed. Amplified DNA may be stockpiled in large quantities, allowing bead arrays to be processed every day of the week, thereby maximizing throughput.
Introduction
Typing single nucleotide polymorphisms (SNPs) is a key method of identifying risk variants associated with disease. Historically, the scope of genotyping experiments has been limited by the technology available. Gel electrophoresis-based genotyping methods are limited in sample- and SNP-throughput[1]. Developing these assays can often be labor-intensive, relying on the makeup and structure of the region surrounding the variant for optimization[1]. TaqMan genotyping assays, developed by Life Technologies, can run a large number of samples quickly and with minimal technician involvement[2], but SNP-multiplexing restrictions continue to limit the total number of genotypes to well under one million per day[3,4]. Sequenom's iPlex platform can also run many samples at once, but, as fewer than one hundred SNPs can be multiplexed together, throughput is comparably low overall[5]. Beckman Coulter's SNP stream technology could theoretically produce over three million genotypes per day, but this technology limits project range to a maximum of only forty-eight SNPs per reaction[4,6]. While the GoldenGate assay can process nearly two hundred DNA samples each day on hundreds or thousands of SNPs per sample, the price per genotype is not competitive with advanced, ultra-high-throughput techniques when typing over three thousand SNPs at once[4,7]. In order to process several million genotypes per day, the scale required for large genome-wide association studies, array-hybridization assays have become the most cost-effective option on the market.
Affymetrix's line of hybridization arrays and Illumina's line of Infinium-based arrays allow potentially hundreds of samples to be typed on hundreds of thousands or millions of SNPs in parallel[4,8]. These SNPs can be scattered across the entire genome, localized in regions of interests, such as exomes, or customized to the user's preference. These arrays have the benefit of not only being able to accurately genotype one million SNPs per sample at once, but also to measure copy number variation, potentially unveiling chromosomal abnormalities. Infinium-aligned OMNI BeadChip arrays currently have the ability to genotype up to nearly five million markers per sample, including half a million custom loci, on up to nearly one hundred samples each day.
As most diseases have a genetic component, these large-scale experiments can be crucial in finding genes associated with disease. High-throughput genotyping allows for efficient genotype generation in sample sets large enough to convincingly detect genetic association at lower minor allele frequencies. Whole-genome genotyping projects can be used to locate regions with statistically significant case-control allele frequency or copy number differences[9,10,11]. According to the National Human Genome Research Institute, genome-wide association studies led to 1,490 separate publications between November 25, 2008 and January 25, 2013, stemming from the discovery of 8,283 SNPs with a p-value less than 1 x 10-5 (see http://www.genome.gov/gwastudies/). These studies, which researched conditions ranging from height to testicular cancer, benefited from the broad approach afforded by a genome-wide analysis. In cases such as these, entire regions of interest might have escaped notice had the scope of typing been too restrictive. Thus, for large-scale association analyses, a genome-wide genotyping technique is the technique of choice.
Different versions of the Infinium assay exist, each intended for use with specific types of arrays. The InfiniumUltra assay, discussed in depth below, is appropriate for many 12- or 24-sample array chips. These often genotype over a hundred thousand SNPs per DNA sample and focus on targeted regions, such as on exome or custom panels. Other assay versions might be required for other chip types, such as the whole-genome genotyping arrays. However, as all Infinium assays share a common basis and mainly differ only by the reagent names, the reagent volumes, or the exact staining reagent procedure, techniques perfected on one assay version can often be universally applied. Other arrays, such as methylation arrays, might use a nearly-identical protocol, as well. Care must be taken to only use the version of the assay required for the chip type in use. Some types, such as ones measuring gene expression level, might require use of a nonInfinium protocol.
Samples must be processed in batches. For example, with the InfiniumUltra assay, prehybridization reagent tubes contain enough volume to run 96 samples, and the tubes cannot be refrozen. Therefore, samples must be run in batches of 96 samples at a time. The samples will be amplified on the first day. After approximately 1 hr of benchwork, the samples must be heated in a convection oven for 20-24 hr. The following day, nearly 4 hr will be spent fragmenting, precipitating, and resuspending the samples, at which point the samples can either be frozen for future use or hybridized to the chip. Loading chips takes nearly 2 hr, after which the samples will be hybridized overnight for 16-24 hr. On the third day, the staining and extension step takes ~4 hr. A further hour will be spent washing, coating, and drying the chips. Finally, the arrays are scanned, which may take from 15-60 min/chip, depending on the type used.
Standard laboratory safety and cleanliness precautions apply. Though the amplification is not PCR-based, separate workstations for pre- and postamplification procedures are necessary in order to minimize likelihood of contamination. The identification number of every kit-supplied reagent in use must be logged on a tracking sheet. Reagents should be thawed immediately before use and inverted several times before dispensing. The DNA to be typed must be high-quality genomic DNA (260/280 absorbance ratio of 1.6-2.0, 260/230 absorbance ratio of below 3.0), isolated by standard methods and quantified with a fluorometer. Degradation of DNA is often a contributing factor in low-quality assay results. Typically, 200 ng of DNA is required, though this amount may vary for some chip types. A Tecan liquid-handling robot can automate many steps of the protocol and minimize human error as a factor.
Protocol
Day One
1. Preparation
- Dispense 200 ng of DNA into a deep-well, 96-sample plate. At least 96 samples (full plates) must be plated to ensure no reagent will be wasted. Label the plate with a barcode sticker supplied by the kit and centrifuge it.
- In order to normalize the volumes, leave the samples in a drawer or fume hood overnight to evaporate the liquid. Cover the plate loosely with a lid or paper towel to keep out dust.
2. Amplification
WARNING: samples should not undergo amplification unless 4 hr will be available on the following day for the fragmentation, precipitation, and resuspension steps.
- Remove the company-supplied box or pack of tubes labeled "Pre" from the -20 °C freezer (one tube of MA1, MA2, and MSM is sufficient for each set of 96 samples). Set tubes of MA2 and MSM on the bench to thaw. Turn on properly-calibrated oven and set to 37 °C.
- Using a reagent basin and a 10 µl, 8-channel pipette, dispense 4 µl of DNA Resuspension Buffer into each well to rehydrate the samples. It is not necessary to discard pipette tips between each column if care is taken not to touch liquid.
- With an 8-channel pipette, dispense 20 µl of MA1 into each well of the plate. For each new reagent, use a fresh reagent basin. Cover the plate with a reusable seal, pulse-centrifuge, and vortex for 1 min at 1,600 rpm on a microplate shaker.
- 2.4) Incubate at room temperature for at least 30 min.
- Dispense 4 µl of 0.1 N NaOH into each well of the plate. Cover the plate with the reusable seal, pulse-centrifuge, and vortex for 1 min at 1,600 rpm.
- Incubate at room temperature for 10 min.
- Dispense 34 µl of MA2 into each well of the plate.
- Dispense 38 µl of MSM into each well of the plate. Cover the plate with the reusable seal, pulse-centrifuge, and vortex for 1 min at 1,600 rpm.
- Place in oven for 20-24 hr.
Day Two
3. Fragmentation
- Remove FMS tubes from the "Post-1" -20 °C box (one tube is sufficient for each set of 96 samples). Thaw on bench or in a room-temperature water bath. After inserting the MIDI-plate insert into a tabletop microsample incubation system, turn on the heat block and set to 37 °C.
- Once tubes are thawed, remove sample plate from oven and pulse-centrifuge. Dispense 25 µl of FMS into each well of the DNA plate. Replace the lid, pulse-centrifuge, and vortex at 1,600 rpm for 1 min.
- Incubate plate in heat block for 1 hr.
4. Precipitation
- Remove PM1 tube from "Post-3" 4 °C box or pack and warm to room temperature (one tube is sufficient for each set of 96 samples). Remove plate from heat block and pulse-centrifuge.
- Dispense 50 µl of PM1 into each well of the plate. Replace the lid, pulse-centrifuge, and vortex at 1,600 rpm for 1 min.
- Incubate plate in heat block for 5 min.
- Remove plate from heat block, turn it off, and discard the plate lid. Dispense 155 µl of 100% isopropanol into each well of the plate. Cover tightly with a new lid and manually invert the plate multiple times to mix.
- Place plate in 4 °C for a minimum of 30 min. Turn on refrigerated centrifuge and set to 4 °C to cool.
- Balance the plate and centrifuge at 4 °C for 20 min at 3,000 x g.
- Inspect the bottom of the plate without inverting it and confirm the samples are precipitated in a blue pellet. If no pellets can be seen, centrifuge again. Get paper towels.
- Discard the lid and quickly remove the liquid by inverting the plate and tapping it forcefully on the benchtop covered in paper towels. Once the plate is inverted, take care not to revert it while any liquid remains. Repeatedly tap the plate against the benchtop until all liquid is removed.
- Set the plate on a test tube rack, inverted, to dry. Incubate at room temperature for 1 hr.
5. Resuspension
- Remove RA1 from "Post-2" -20 °C box and thaw in room temperature water bath. Turn on the oven and set it to 48 °C.
- Once thawed, dispense 23 µl of RA1 to each well of the sample plate. Do not pour the entire bottle of RA1 into the basin; save 30 ml for later. If the samples will be hybridized onto the arrays later that day, place the RA1 into a 4 °C cooler. If the samples will be run at a later date, label the bottle and refreeze the RA1.
- Heat-seal a new lid onto the plate. Pulse-centrifuge the plate and place it in the oven for 1 hr.
- Remove the plate from the oven and vortex at 1,800 rpm for 1 min.
The samples can safely be held at this stage for up to one week. Plates may be stockpiled, and samples may be reorganized to prepare for the bead chip application, if necessary. If proceeding with the procedure, leave the plate at room temperature and turn on the heat block. Otherwise, store the plate at -20 °C.
6. Hybridization
WARNING: Samples should not undergo hybridization unless 5.5 hr are available on the following day for the staining and wash steps.
- Turn on the heat block and set it to 95 °C. Turn on the oven and set it to 48 °C.
- Once the temperature has stabilized, incubate the plate on the heat block for 20 min.
- While the plate is denaturing, remove the box of bead chips from 4 °C and set it on the bench. Take a bottle of XC4 (from room-temperature kit) and add 330 ml of 100% ethanol. Shake it well and incubate at room temperature overnight. Prepare formamide/EDTA mix (95% formamide, 0.2% EDTA (0.5 M), 4.8% H2O by volume) and freeze in separate 15 ml increments. Excess formamide may be stockpiled.
- Remove the sample plate from the heat block. Incubate at room temperature for 30 min.
- While the plate is cooling, prepare the hyb chamber. One chamber will be needed for every four bead chips processed.
Place a rubber mat on the top of the hyb chamber base, aligning the thicker hole with the front of the base. The barcode symbol etched into the base should still be visible (see Figure 2). - Using a 1,000 µl pipette, dispense 400 µl of PB2 into each of the eight humidifying reservoirs carved into the base. PB2 can be found in "Post-3" box or pack. Place the hyb chamber lid on the base and clasp both ends by closing two clasps on diagonally opposite sides first.
- Remove the individual silver packs of bead chips from their respective boxes, but, in order to minimize the chips' exposure to air and light, do not yet open them. Carefully scan the barcodes of the chips and record the order in which they will be loaded on a tracking sheet, along with the box IDs from which they came. A thorough understanding of where each individual DNA sample will be dispensed on the bead chips is necessary.
- Remove the individual silver packs of bead chips from their respective boxes, but, in order to minimize the chips' exposure to air and light, do not yet open them. Carefully scan the barcodes of the chips and record the order in which they will be loaded on a tracking sheet, along with the box IDs from which they came. A thorough understanding of where each individual DNA sample will be dispensed on the bead chips is necessary.
- Just before the 30 min cool down has completed, open the silver bead chip packs. Remove the chips from their clear plastic sleeves. Without touching the beads, place every bead chip on a hyb chamber insert, orienting the chip barcode with the barcode symbol etched into the insert's top surface.
- Peel off and discard the lid of the sample plate. Take 15 µl of sample from the sample plate and slowly dispense on the inlet to the array (see Figure 3). Extraordinary care must be taken to place the correct sample on the correct bead chip in the correct position, matching the bead chip order recorded earlier. A multichannel pipette may be used to dispense liquid on the chips, but forethought must be taken to aspirate only the number of samples that can be safely placed on the bead chip, as the number of rows on a plate does not always match the number of rows on a chip.
- Once every sample has been placed on its corresponding array, visually inspect the chip for bubbles or regions not coated in liquid. If these problems exist, gently rock the insert. If necessary, more liquid may be added to array. Take care to add the correct DNA sample.
- Open the hyb chamber and place the inserts over the humidifying reservoirs. The barcode of the chip should be placed over the barcode etched into the hyb chamber base. Replace the hyb chamber cover and close all four clasps by closing the clasps on diagonally opposite sides first.
- Incubate the hyb chamber in the oven for 16-24 hr. Take care not to tilt the chambers when moving them.
- If more than one plate is to be processed, return to step 6.2. Processing more than 24 bead chips in a day is not recommended in order to both minimize the likelihood of human error when handling a large quantity of consumables or chips and to minimize the amount of time required to process the chips in future steps, as care must be taken to limit their exposure to air as much as possible. One bottle of XC4 is sufficient for up to 24 bead chips.
Day Three
7. Staining Preparation
- Remove the hyb chambers from the oven and incubate at room temperature for 25 min. If processing more than two hyb chambers, stagger their dual removal every 10 min. In order to keep the chips from drying, do not yet open the hyb chambers.
- While the hyb chambers are cooling, remove tubes of XC1, XC2, TEM, STM, and ATM from the "Post-1" -20 °C box and set on the bench to thaw. Take a bottle of RA1 from the "Post-2" box at -20 °C (or 4 °C if reusing the reagent from the previous day) and thaw in a room temperature water bath. Thaw a tube of 95% formamide as well. For 8 bead chips processed, two tubes of each reagent, 10 ml RA1, 15 ml formamide, and 150 ml XC3 (found in a room-temperature, company-supplied box) will be required.
- For every chip processed, one glass slide, one plastic flow-through brace, two metal clasps, and one plastic spacer will be required. For the plastic spacer, use only the clear one; separate and discard the opaque spacer. In addition, two bead chip wash dishes and one bead chip tray will be needed, along with one liquid assembly station and one plastic assembly bar.
- Clean the glass slides by spraying them with ethanol and wiping them dry.
- Turn on the hot water bath that's attached to the flow-through chamber rack and set it to 44 °C. Gently shake the chamber rack to dislodge any bubbles.
- When the hyb chamber has cooled for 25 min, fill a wash dish with PB1 (approximately 200 ml). PB1 can be found in the "Post-4" room temperature box.
- Open one hyb chamber. Remove the cover seal from a bead chip by grasping the seal at one corner and gently peeling diagonally (see Figure 4). Once the seal is removed, immediately place the chip in a bead chip tray and submerge in PB1 without touching the beads. Repeat until the tray is filled with bead chips, taking care not to let any chip dry.
- Gently agitate the tray to remove any bubbles and leave the chips submerged for 1 min. Fill a second wash dish with PB1. Agitate the tray again.
- Move the tray of bead chips into the second wash dish. Gently agitate and let soak for 1 min, then agitate again.
- In the liquid flow-through assembly station, place four black plastic flow-through braces in the grooves. Fill the station with PB1 until the liquid nearly reaches the height of the eight assembly brackets (approximately 150 ml).
- Remove a bead chip from the tray and place it into the assembly station on a plastic flow-through brace. The chip barcode should be aligned above the barcode symbol etched into the assembly station. Repeat for the other three chips that can fit within the assembly station.
- Place a clear plastic spacer on top of a bead chip. The outer edges of the spacer should surround the brackets within the assembly station. Repeat for the other chips submerged in the PB1.
- Place the plastic assembly bar in the assembly station by fitting it over the grooves. Gently place a glass slide on top of the plastic spacer by pushing the rear end of the slide against the assembly bar and slowly lowering the front into the liquid. The groove on the top of the slide should face downwards, directly above the barcode of the chip, leaving a gap between the bead chip and the slide at the barcode end. Repeat for the other chips submerged in the PB1. For a complete flow-through assembly diagram, see Figure 5.
- Check for bubbles between the chip and slide. If bubbles appear, gently raise the slide at the barcode end and try again. Persistent bubbles can be wiped from the glass with a paper laboratory towel (Kimwipe).
- Snap two metal clasps around the glass slide, one towards the barcode end and one towards the rear. The edges of the clasps should grip the plastic flow-through brace under the chip. Repeat for every chip submerged in PB1.
- Remove the completed flow-through assemblies and place horizontally on the bench. Take care not to tip the assembly vertically, allowing the liquid under the glass slide to escape. If more chips are waiting in the wash tray, they can be assembled under the same PB1. If other chips are waiting in their original hyb chambers, discard all liquid in the assembly station and wash dishes, and return to step 7.6.
- Once all bead chips are in their flow-through assemblies, take a pair of surgical scissors and cut both ends of the spacer off, as near to the glass slide as possible.
8. Staining and Extension
- Verify that the flow-through chamber rack has reached 44 °C in multiple positions with a temperature probe. If the temperature is off by more than half a degree, adjust the temperature of the water bath.
- Place a bead chip flow-through assembly on the chamber rack by sliding the assembly down and hooking the brace at the top. The back side of the bead chip should be touching the chamber rack. The glass slide should be facing out, with the groove at the top to form a reagent reservoir. Repeat for every bead chip assembly.
- Using a 200 µl pipette, add 150 µl RA1to the flow-through assemblies by dispensing the liquid into the glass reservoir. Incubate for 30 sec. Repeat 5x.
- Using a 1,000 µl pipette, add 450 µl XC1to the flow-through assemblies by dispensing the liquid into the glass reservoir. Incubate for 10 min.
- Add 450 µl XC2 to the flow-through assemblies by dispensing the liquid into the glass reservoir. Incubate for 10 min.
- Add 200 µl TEM to the flow-through assemblies by dispensing the liquid into the glass reservoir. Incubate for 15 min.
- Add 450 µl 95% formamide/EDTA mix to the flow-through assemblies by dispensing the liquid into the glass reservoir. Incubate for 1 min. Repeat 1x.
- Incubate the flow-through assemblies for 5 min.
- Set the hot water bath to the temperature listed on the tubes of STM (or 37 °C if none is shown). Make sure each STM tube has the same temperature listed.
- Add 450 µl XC3 to the flow-through assemblies. Incubate for 1 min. Repeat 1x.
- When the hot water bath temperature has reached the desired temperature, add 250 µLSTM to the flow-through assemblies by dispensing the liquid into the glass reservoir. Incubate for 10 min.
- Add 450 µl XC3 to the flow-through assemblies by dispensing the liquid into the glass reservoir. Incubate for 1 min. Repeat 1x.
- Incubate the flow-through assemblies for 5 min.
- Add 250 µl ATM to the flow-through assemblies by dispensing the liquid into the glass reservoir. Incubate for 10 min.
- Add 450 µl XC3 to the flow-through assemblies by dispensing the liquid into the glass reservoir. Incubate for 1 min. Repeat 1x.
- Incubate the flow-through assemblies for 5 min.
- Add 250 µl STM to the flow-through assemblies by dispensing the liquid into the glass reservoir. Incubate for 10 min.
- Add4 50 µl XC3 to the flow-through assemblies by dispensing the liquid into the glass reservoir. Incubate for 1 min. Repeat 1x.
- Incubate the flow-through assemblies for 5 min.
- Repeat steps 8.14 to 8.19 1x.
- Remove the flow-through assemblies from the chamber rack and turn off the water bath.
9. Washing and Sealing
- Fill a staining bead chip wash dish with PB1 (approximately 315 ml). Two vertical wash dishes and one vertical bead chip tray will be needed, along with at least one vacuum manifold.
- Disassemble a flow-through assembly by inserting a thin metal bar between the clasps and the brace and then pivoting. Set aside the glass slide and remove the bead chip. Immediately place the bead chip in the vertical bead chip tray and submerge in PB1. Repeat for every flow-through assembly, taking care to face each bead chip in the same direction and minimizing their exposure to air.
- Gently agitate the bead chip tray to remove bubbles. Incubate the submerged bead chips in PB1 for 5 min. Fill the second vertical wash dish with the XC4 prepared the previous day. Agitate the bead chip tray again.
- Move the bead chip tray to the wash dish filled with XC4. Gently agitate the tray to remove bubbles. Incubate for 5 min and agitate again.
- In one smooth motion, pull the bead chip tray from the wash dish and set on a test tube rack, so that the beads on every bead chip face upwards. With self-locking tweezers, slide a bead chip out of the tray and place on a test tube rack. Repeat for every bead chip.
- Carefully transfer the test tube rack of chips to a vacuum desiccator. Close and turn on the vacuum, ensuring a proper seal. Incubate for 50-55 min. If necessary, warm up the scanner during incubation.
10. Scanning
- Turn off the vacuum and slowly return the pressure to atmospheric. Check that the bead chips are dry. If necessary, wipe the edges and bottom of the chip with a paper towel to remove any liquid or debris.
- Activate the scanning software and move bead chips to scanning tray. Download the chip's decode files (dmaps) by activating the Decode File Client software and inputting the desired bead chip barcodes, along with their corresponding box IDs. Unscanned bead chips may safely be stored in a dry, dark area for up to 72 hr.
- Once the chips are properly seated in the tray and the decode files are recognized by the software, start scanning.
Representative Results
A properly-processed bead chip should display bright and distinct red and green laser-light intensities while scanning. In Figure 6, the iScan scanning software displays a standard genomic DNA sample successfully hybridized to a custom-panel SNP genotyping array. The labeled nucleotides that were attached during the extension and staining steps fluoresce under the lights of the two lasers. As these nucleotides selectively extend the bead's oligonucleotide chain hybridized to the fragmented DNA strand, and as the oligonucleotide chains are designed to terminate at the site of the variant, the color and intensity of the resulting signal can be used to determine the alleles present at the SNP site.
A user-generated sample sheet, which matches sample IDs and clinical information to chipID and position, and an array-specific SNP manifest, obtained directly from the company, are both necessary in order to import the scanner output files into the GenomeStudio analysis software. Example sample sheets can be found on the company's website. Once the GenomeStudio project is built, genotypes can be obtained from the resulting intensity clusters automatically generated by the software. Though multiple display options exist, the default view, Norm-R (a normalized intensity value) vs. Norm-Theta (an allelic intensity ratio), is often the easiest plot to differentiate three distinct clusters. Most SNPs should show one, two, or three clusters, depending on the minor allele frequency.
The three clusters represent samples demonstrating AA, AB, or BB alleles (see Figure 7). These clusters can be assigned genotypes either by importing a standard clustering position template directly from the company, by selecting "Cluster All SNPs" from the Analysis toolbar of the software, or by clicking and dragging the colored circles on the intensity plots to manually edit calls. The color of the sample on the intensity plot (red, purple, or blue) indicates the call (AA, AB, or BB, respectively); black indicates no call. By scrolling through the SNPs listed in the Full Data pane of the software, the clustering for each SNP can be viewed or recalled. Once the clusters are assigned satisfactory calls, the Full Data pane of the software lists the genotypes for each individual sample at each particular SNP. The Column Chooser option above the table can toggle data formats.
Data can be exported either through the Analysis toolbar or directly from the Full Data table for in-depth analysis.
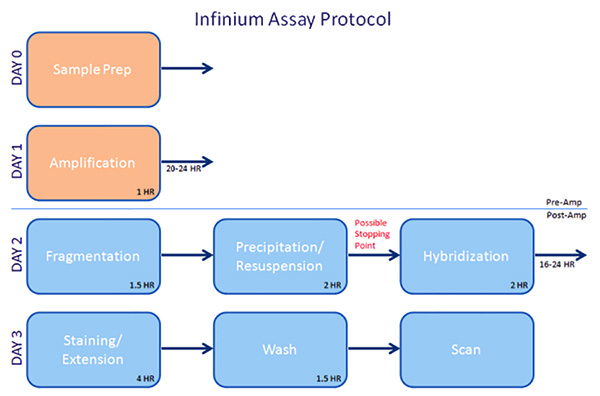
Figure 1. Overview – Infinium Assay Protocol. Click here to view larger image.

Figure 2. Complete Hyb Chamber base and mat, plus lid.[12] Click here to view larger image.
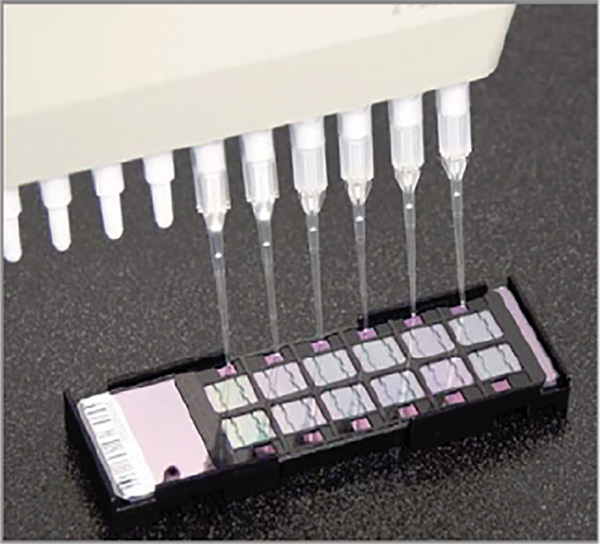
Figure 3. Loading a Beadchip – Dispense the sample on the inlet ports.[12] Click here to view larger image.

Figure 4. Removing the Beadchip Coverseal – Grasp the seal at the corner and gently peel diagonally.[12] Click here to view larger image.

Figure 5. Complete Beadchip Flow-through Assembly – The BeadChip is separated from a glass slide with a spacer and bound with clasps. Click here to view larger image.
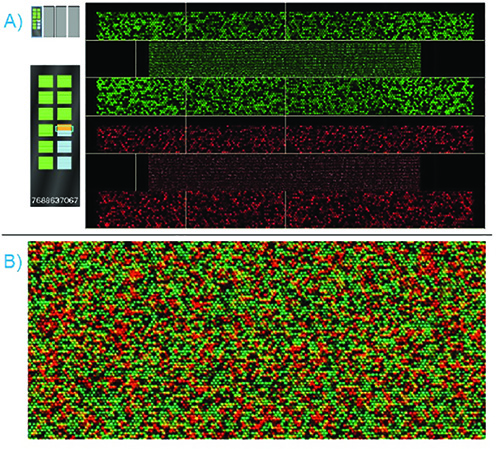
Figure 6. Successful BeadChip Scan – A) The BeadChip is scanned with both a red and green laser; the scanning software displays both simultaneously. Sections passing intensity QC will highlight green on the BeadChip display to the left. Sections failing intensity QC will highlight red on the BeadChip display. B) Once scanning is complete, the software will overlay the red and green displays. A zoomed-in image is shown. The color and intensity of each individual bead indicates the allele present. Click here to view larger image.
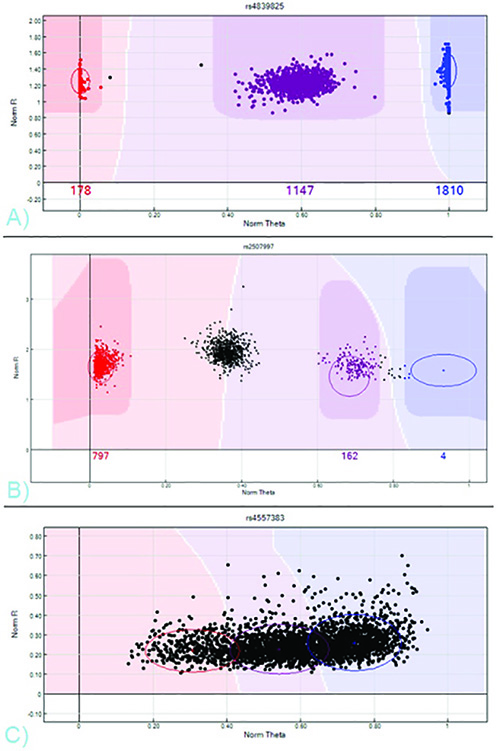
Figure 7. SNP NORM-R vs. NORM-THETA Cluster Profiles – A) A valid SNP with three distinct clusters representing AA, AB, and BB genotypes, colored red, purple, and blue. B) A SNP requiring editing. The middle cluster, which should be homozygous AB, is left un-called. The BB cluster is mistakenly called AB. C) A poor-performing SNP. No genotypes can be obtained from this intensity plot, as no distinct clusters exist. Click here to view larger image.
Discussion
Large-scale genotyping applications have been used to better understand the genetic mechanism underlying many human diseases. The discovery of any significant variant through a genome-wide association analysis can flag a candidate region for further study. In addition, genotype data is a good tool for quality control on sequencing projects.
To maximize sample throughput, multiple sample plates can be amplified and stored in their fragmented, resuspended states. Eight plates may be amplified in a single day, combining the first 24 hr of the protocol for multiple batches and providing enough material for ~2-8 days of chip-processing. If amplified plates are stockpiled beforehand, and if new samples are hybridized to chips immediately after scanning begins on the previous run, processing can run continuously without the need to pause for additional sample preparation. Therefore, though samples will take three days to undergo the complete assay, data can be generated daily. Assuming24 chips are processed every day, a 5 day workweek allows for over a 1,000 DNA samples to be run on a 12-sample bead chip. If any step or reagent has failed, however, multiple batches might be at risk for poor performance before any correction can be applied. Errors might escape notice until the arrays are scanned or analyzed; therefore, if throughput is maximized, hundreds of samples in various stages of the protocol might already have received the same defective treatment upon discovery. As lost reagents and data cannot be recovered, the user must weigh these risks against the need for an accelerated workflow.
The GenomeStudio analysis software is the first chance to truly gauge the success of the genotyping process. If the Norm-R vs. Norm-Theta intensity plots are properly clustered, the average call rate (percent of total SNPs successfully typed) of the samples should approach 99%, though this value varies slightly depending on array type. The data from any sample with a call rate lower than 85-90% is not trustworthy and should be discarded. For quality control purposes, results should be compared to any previously-known genotypes whenever possible. If no such data exists, intentional sample duplication is a useful tool in verifying plate or array placements. These duplicate pairs should be placed on separate chips, plates, batches, or projects; their genotypes checked upon generation. While specific QC constraints vary according to the investigator's preference, common SNP constraints are based on sample call success, Hardy-Weinberg equilibrium, or missingness between cases and controls, while common sample constraints are based on call rates, Mendelian inconsistencies, or cross-references of X-chromosome heterozygosity to clinical gender data[13].
If any problem arises, the Controls Dashboard, found in the analysis suite, can be submitted to the company in order to determine cause. These controls can often narrow down the issue to the likeliest step or reagent failure. If any SNPs of interest are found through an Infinium genotyping experiment, their intensity plots should be double-checked in GenomeStudio for clustering errors before further research is conducted.
A failed Infinium genotyping experiment is likely due to human processing error or poor-quality input DNA. Sample quantification must be accurate and precise. For best results, any reagents added to any sample or chip must be dispensed at the volume set by the protocol. Pipettes must be properly calibrated. Reagents should not be run after expiration and should not be refrozen once thawed, save for the RA1 reagent. In order to minimize possible staining and extension errors, the formamide/EDTA mixture should be prepared fresh every month. All -20 °C reagents should be stored in manual-defrost freezers only. All labware used in the staining, extension, and wash portions of the protocol should be rinsed thoroughly with water and mild detergent immediately upon disuse. The humidifying reservoirs in the hyb chamber should be scrubbed with a test tube brush and mild detergent. The glass slides should be washed with 10% bleach, as instructed by their user manuals, once a week.
Disclosures
The authors have nothing to disclose.
Acknowledgements
Funding for this work has been provided by NIH P20 GM103456, NIH RC2 AR058959, and NIH R56 AI063274
Materials
| Consumable or Equipment | Manufacturer | Part Number | Minimum Required for 96 Samples |
| 0.8 ml Deep Well Plate | Thermo Scientific | AB-0765 | 1 |
| Plate Mats | Thermo Scientific | AB-0674 | 2 |
| Reagent Basin | Fisher Scientific | 13-681-502 | 9 |
| Heat-seal Sheets | Thermo Scientific | AB-0559 | 1 |
| Flow-through Spacer | Fisher Scientific | NC9563984 | 6 |
| Pipette tips – 200 μl | Rainin | GP-L200F | 192 |
| Pipette tips – 10 μl | Rainin | GP-L10F | 16 |
| Pipette tips – 1,000 μl | Rainin | GP-L1000F | 16 |
| DNA Suspension Buffer | Teknova | T0220 | 0.5 ml |
| 0.1 N NaOH | Fisher Scientific | AC12419-0010 | 0.5 ml |
| Isopropanol (HPLC grade) | Fisher Scientific | A451 | 15 ml |
| Ethanol (200-proof) | Sigma-Aldrich | 459836 | 330 ml |
| Formamide (100%) | Thomas Scientific | C001K38 | 15 ml |
| EDTA (0.5 M) | Amresco | E177 | 0.2 ml |
| 10 μl 8-channel Pipette | Rainin | L8-10XLS | 1 |
| 200 μl 8-channel Pipette | Rainin | L8-200XLS | 2 |
| 1,000 μl Single-channel Pipette | Rainin | L-1000XLS | 1 |
| Microplate Shaker | VWR | 13500-890 | 1 |
| Refrigerated Microplate Centrifuge | VWR | BK369434 | 1 |
| Hybridization Oven | Illumina | SE-901-1001 | 1 |
| Hybex Microsample Incubator | SciGene | 1057-30-0 | 1 |
| Hybex MIDI Heat Block Insert | Illumina | BD-60-601 | 1 |
| Heat Sealer | Thermo Scientific | AB-0384 | 1 |
| Hyb Chamber w/ Insert and Mat | Illumina | BD-60-402 | 2 |
| Surgical Scissors | Fisher Scientific | 13-804-20 | 1 |
| Flow Through Assembly Parts | Illumina | WG-10-202 | 8 |
| Wash Rack and Dish | Illumina | BD-60-450 | 1 |
| Genepaint Chamber Rack | Tecan | 760-800 | 1 |
| Temperature Probe | Illumina | A1-99-109 | 1 |
| Staining Rack and Dish | Illumina | WG-10-207 | 1 |
| Self-Closing Tweezers | Ted Pella, Inc | 5374-NM | 1 |
| Vacuum Manifold | Ted Pella, Inc | 2240 | 1 |
| iScan or HiScan | Illumina | – | 1 |
References
- Shi, M. M. Enabling large-scale pharmacogenetic studies by high-throughput mutation detection and genotyping technologies. Clin. Chem. 47 (2), 164-172 (2001).
- Livak, K. J. SNP genotyping by the 5′-nuclease reaction. Methods Mol. Biol. 212, 129-148 (2003).
- Seeb, J. E., Pascal, C. E., Ramakrishnan, R., Seeb, L. W. SNP genotyping by the 5′-nuclease reaction: advances in high throughput genotyping with non-model organisms. Methods in Mol. Biol. 578, 277-292 (2009).
- Bayés, M., Gut, I. G., ed, . Overview of Genotyping. Molecular Analysis and Genome Discovery. , 1-23 (2011).
- Lee, Y., Seifert, S. N., Fornadel, C. M., Norris, D. E., Lanzaro, G. C. Single-Nucleotide Polymorphisms for High-Throughput Genotyping of Anopheles arabiensis in East and Southern Africa. J. Med. Entomol. 49 (2), 307-315 (2012).
- Liu, Z. SNP Genotyping Platforms. Next generation sequencing and whole genome selection in aquaculture. , 123-132 (2011).
- Shen, R., Fan, J. B., et al. High-throughput SNP genotyping on universal bead arrays. Mutat. Res./Fundam and Mol. Mech. of Mutagenesis. 573 (1), 70-82 (2005).
- Ragoussis, J. Genotyping technologies for all. Drug Discov. Today: Technol. 3 (2), 115-122 (2006).
- Wu, C. C., Shete, S., Jo, E. J., et al. Whole-Genome Detection of Disease-Associated Deletions or Excess Homozygosity in a Case-Control Study of Rheumatoid Arthritis. Hum. Mol. Genet. , (2012).
- Green, E. K., Hamshere, M., Forty, L., et al. Replication of bipolar disorder susceptibility alleles and identification of two novel genome-wide significant associations in a new bipolar disorder case-control sample. Mol. Psychiatry. , (2012).
- Shete, S., Hosking, F. J., Robertson, L. B., et al. Genome-wide association study identifies five susceptibility loci for glioma. Nat. Genet. 41 (8), 899-904 (2009).
- Inc Illumina. . Infinium HD Ultra User Guide 11328087 RevB-1. , 15-101 (2009).
- Turner, S., Armstrong, L. o. r. e. n. L., Yuki Bradford, ., et al. Quality Control Procedures for Genome Wide Association Studies. Curr Protoc Hum Genet. , 1-19 (2011).
