The protocols outlined were used to examine survival of N. gonorrhoeae after exposure to primary human neutrophils 5,26. Neutrophils were infected with N. gonorrhoeae and processed with protocol 1, using the green-fluorescent viability dye SYTO9 and the red-fluorescent propidium iodide (Figure 4A). The dyes were added in the presence of saponin, which sequesters cholesterol to preferentially permeabilize host cell plasma membranes, not N. gonorrhoeae membranes. Other detergents may need to be tested if the membranes of the bacteria of interest contain high amounts of cholesterol. All N. gonorrhoeae stain with SYTO9, but only bacteria with compromised membranes stain with propidium iodide. The propidium iodide overcomes the SYTO9 fluorescence, so live bacteria appear green and dead bacteria appear red 27. In some bacterial species, propidium iodide may not completely overcome SYTO9 staining, resulting in dead bacteria appearing yellow or orange. The ratio of propidium iodide to SYTO 9 may need to be optimized for different bacterial species. Prior to permeabilizing the cells and adding SYTO9 and propidium iodide, an Alexa Fluor 647-coupled soybean lectin was added to the infected cells to detect extracellular N. gonorrhoeae, and the far-red fluorescence signal was false-colored blue. Thus, in this protocol external viable bacteria appear turquoise (blue + green) and external nonviable appear magenta (blue + red). Internal viable bacteria appear green only (arrow), and internal nonviable bacteria appear red only (arrowhead). It is important to note that in infected cells, the eukaryotic cell nucleus will be stained by SYTO9 and propidium iodide ("N", Figure 4A). The external viable, external nonviable, internal viable, and internal nonviable bacteria were quantified from fluorescence images to yield the percent of internalized bacteria and the percent viable bacteria inside and outside host cells.
Figure 4B is a representative image of N. gonorrhoeae-infected neutrophils processed with protocol 2, using the viability dyes SYTOX Green and DAPI. This protocol avoids the use of propidium iodide, which fluoresces in both red and ultraviolet channels on the fluorescence microscope. All N. gonorrhoeae stain with DAPI (blue), but only bacteria with compromised membranes stain with SYTOX Green. As described above, before permeabilizing the cells, an Alexa Fluor 647-coupled soybean lectin was added to the infected cells to detect extracellular N. gonorrhoeae, whose fluorescence in this case is false-colored red. Therefore, external viable bacteria appear magenta (red + blue) and external nonviable bacteria appear yellow (red + green/blue). Internal viable bacteria appear blue only (arrows), and internal nonviable bacteria appear green/blue (arrowheads). Depending on the relative intensity of DAPI vs. SYTOX Green fluorescence, nonviable bacteria may appear turquoise. Similar to cells processed with propidium iodide and SYTO9, the neutrophil nucleus is stained by SYTOX Green and DAPI in Figure 4B (indicated by "N"). Exposure of infected cells to SYTOX Green and DAPI reveals the percent internalization and viability of bacteria in host cells, quantified as described above for propidium iodide and SYTO9. Results from N. gonorrhoeae-infected neutrophils processed using SYTOX Green and DAPI are comparable to those obtained with propidium iodide and SYTO9 26.
As depicted in Figure 4C, protocol 3 is an adaptation of protocol 2, in which the bacterial viability dyes were combined with immunofluorescence for the primary or azurophilic neutrophil granule protein CD63. This experiment was conducted in order to define the composition of phagosome compartments inside neutrophils that contain viable or nonviable N. gonorrhoeae 26. This protocol requires a microscope for four-color fluorescence capability to detect viable bacteria, nonviable bacteria, extracellular bacteria, and subcellular marker. Since the viability dyes are not compatible with aldehyde fixation (our unpublished observations), we performed immunofluorescence without fixation, using an antibody directly coupled to a fluorophore to minimize the time needed to detect the antigen inside cells. This antibody is coupled to the red fluorophore Alexa Fluor 555. As in protocol 2, viable and nonviable bacteria were distinguished using DAPI and SYTOX Green, respectively, and external bacteria were stained using Alexa 647-coupled soybean lectin. External bacteria fluoresce far-red and are false-colored purple. External viable bacteria appear purple + blue and external nonviable bacteria appear purple + green/blue. Internal viable bacteria appear blue (arrow) and internal nonviable bacteria appear green (arrowhead). CD63-positive granules and phagosomes appear red. The neutrophil nucleus is stained with the DNA specific dyes ("N"). When analyzing these images, each individual bacterium was classified as external vs. internal, viable vs. nonviable, and positive or negative for a ring of CD63 staining. N. gonorrhoeae was considered to reside in a neutrophil CD63-positive phagosome if there is ≥50% of a ring of CD63 fluorescence surrounding the bacteria, and in a CD63-negative phagosome if there is <50% of a ring of CD63 fluorescence surrounding the bacteria. The nonviable intracellular bacterium indicated by the arrowhead in Figure 4C has a ring of staining for CD63 surrounding the bacteria. This ring indicates primary granules are enriched at this phagosome. The viable intracellular bacterium, indicated by the arrow, lacks staining for CD63, indicating primary granules are not enriched at its phagosome. We used this technique to show a correlation between the viability of N. gonorrhoeae and residence in a primary granule-negative phagosome 26. Because this assay tracks both viability and bacterial localization using a protein of interest, it is possible to determine if nonviable and viable bacteria reside in the same or different locations. This information provides important initial insight into mechanisms that contribute to bacterial survival in host cells.
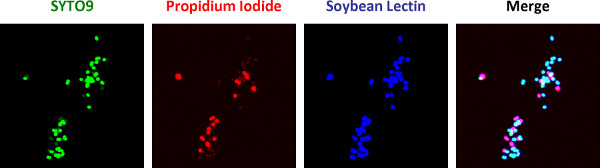
Figure 1. A mix of viable and nonviable N. gonorrhoeae was exposed to the Alexa Fluor 647-coupled soybean lectin, propidium iodide, and SYTO9 according to protocol 1, in the absence of host cells. All bacteria are accessible to the lectin and fluoresce blue. Nonviable bacteria appear blue + red and viable bacteria appear blue + green.
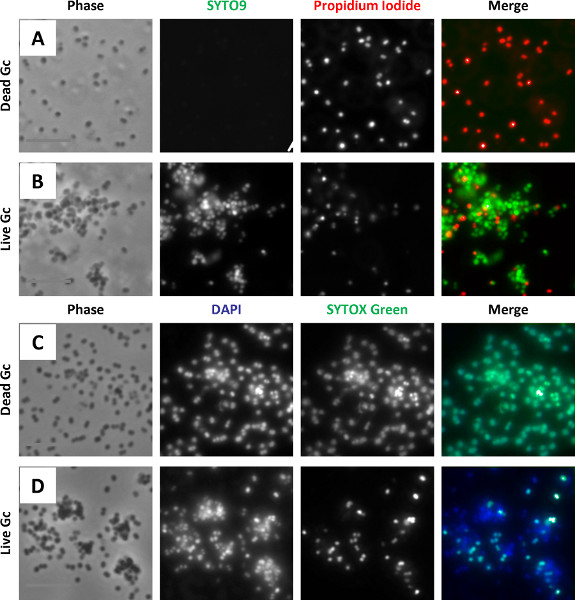
Figure 2. (A-B) Mid-logarithmic phase N. gonorrhoeae was exposed to propidium iodide and SYTO9 as in protocol 1, with (A) or without (B) isopropanol treatment to kill the bacteria. Nonviable bacteria are accessible to propidium iodide and appear red. Viable bacteria are stained with SYTO9 and appear green. (C-D) Mid-logarithmic phase N. gonorrhoeae was exposed to DAPI and SYTOX Green as in protocol 2, with (C) or without (D) isopropanol treatment to kill the bacteria. Nonviable bacteria are accessible to DAPI and SYTOX Green and appear blue + green. Viable bacteria are stained with DAPI only and appear blue only.
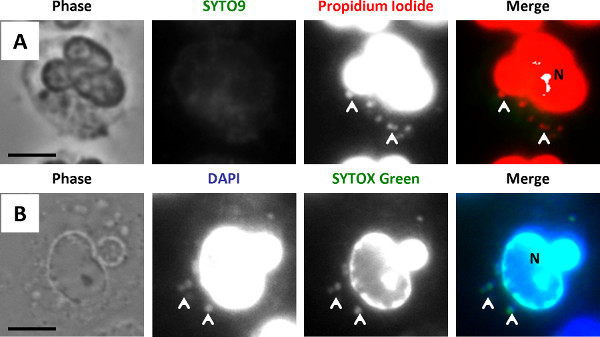
Figure 3. (A) Neutrophils were allowed to bind and internalize isopropanol-killed N. gonorrhoeae, then exposed to propidium iodide and SYTO9 according to protocol 1. Nonviable bacteria are stained with propidium iodide and appear red. As all bacteria are nonviable, no SYTO9 positive, green bacteria are detected. (B) Neutrophils were allowed to bind and internalize isopropanol-killed N. gonorrhoeae, then exposed to DAPI and SYTOX Green according to protocol 2. Nonviable bacteria are stained with DAPI and SYTOX Green and appear blue + green. As all bacteria are nonviable, no DAPI only, blue only bacteria are detected. N labels the neutrophil nucleus. Scale bar = 5 μm. Arrowheads indicate some of the nonviable bacteria.
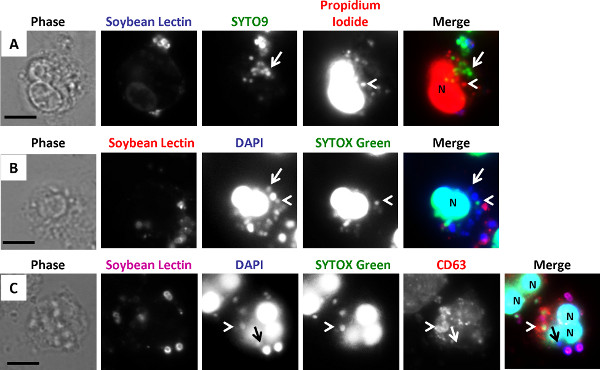
Figure 4. (A) N. gonorrhoeae-infected human neutrophils were exposed to Alexa Fluor 647-coupled soybean lectin (false-colored blue), then permeabilized with saponin in the presence of the viability dyes propidium iodide (red), to label dead bacteria, and SYTO9 (green), to counterstain live bacteria. External nonviable bacteria appear blue + red, internal nonviable bacteria appear red only, external viable bacteria appear blue + green, and internal viable bacteria appear green only. (B) N. gonorrhoeae was labeled with DAPI (blue), then presented to neutrophils. Infected cells were then exposed to Alexa Fluor 647-coupled soybean lectin (false-colored red), then permeabilized with saponin in the presence of SYTOX Green (green) to label dead bacteria. External nonviable bacteria appear red + green, internal nonviable bacteria appear green + blue, external viable bacteria appear red + blue, and internal viable bacteria appear blue only. (C) N. gonorrhoeae-infected neutrophils were processed as in B, except an Alexa Fluor 555-coupled antibody against the neutrophil primary granule protein CD63 (red) was added at the time of permeabilization. The Alexa Fluor 647-soybean lectin staining was false-colored purple. External nonviable bacteria appear purple + green, internal nonviable bacteria appear green only, external viable bacteria appear purple + blue, internal viable bacteria appear blue only, and CD63 staining appears red. The inset shows the CD63 fluorescence of the area of the image indicated with the white square. In A-C, N labels the neutrophil nucleus. Scale bar = 5 μm. Arrowheads indicate nonviable bacteria and arrows indicate viable bacteria.
