Die Anzahl von Kristallstrukturen von Proteinen und Proteinkomplexen schnell erhöht in den letzten Jahren. Sie präsentieren unschätzbare Schnappschüsse von der strukturellen Organisation dieser Proteine und eine Grundlage für die Struktur-Funktions-Analyse. Jedoch die Dynamik von Proteinen und Konformationsänderungen, die für ihre Funktionen sind, werden selten durch Röntgenkristallographie ergab. Cryo-Elektronenmikroskopie, auf der anderen Seite, ist in der Lage, Protein-und Protein-Komplexe in verschiedenen Konformationen zu erfassen, aber im Allgemeinen können Konformationsänderungen nicht lösen auf Sekundärstrukturebene ein. Konformationsdynamik von Proteinen in Lösung in atomarer Informationen sind nur für NMR gelöst werden, aber dieses Verfahren ist immer noch relativ kleine Proteine von Größen (in der Regel ≤ 30 kDa) beschränkt und erfordert hohe Konzentrationen von Proteinen (≥ 100 &mgr; M), die Experimente mit behindert Oligomerisierung oder Aggregation neigen Proteine 2. Eine Methode, dieder Lage ist, zwischen hochauflösende Röntgen-Kristallographie und Kryo-Elektronenmikroskopie und die Brücke nicht durch Größe oder Protein-Konzentration begrenzt ist Amid-Wasserstoff-1 H / 2 H-Austausch (HX) in Kombination mit der Massenspektrometrie (MS). In den letzten Jahren hat dieses Verfahren einen wertvollen analytisches Werkzeug für die Analyse von Proteindynamik, Proteinfaltung, der Proteinstabilität und Konformationsänderungen 3-5 entwickelt. Die molekulare Grundlage dieses Verfahrens ist die Labilität Rückgrat Amidwasserstoffe in Proteinen, die durch Deuteriumatome austauscht, wenn das Protein in D 2 O-Lösung gegeben. Die anschließende Erhöhung der Proteinmasse im Laufe der Zeit wird mit hochauflösenden MS gemessen.
Kurz unstrukturierten Peptiden HX hängt nur von der Temperatur, Katalysatorkonzentration (OH -, H 3 O +, dh pH-Wert, siehe Abbildung 3) und Aminosäure-Seitenketten der benachbarten Resten durch induktive, Katzekatalytische und sterische Effekte. Diese Auswirkungen auf die intrinsische chemischen Austauschrate k ch wurden elegant von Bai et al. 6 quantifiziert und ein Programm zur Verfügung (mit freundlicher Genehmigung Z. Zhang), die k ch für jede Aminosäure in einem Polypeptid abhängig von pH-Wert und Temperatur berechnet. Bei neutralem pH-Wert und Umgebungstemperaturen ch k in der Größenordnung von 10 1 bis 10 3 sec -1. Ionen in das Innere einer eng gefalteten Protein – in gefalteter Proteine HX 2-9 Größenordnungen langsamer hauptsächlich auf Wasserstoffbrückenbindung in der Sekundärstruktur und aufgrund eingeschränkter Zugangs hydratisierter OH geringem Ausmaß sein. HX in nativen Proteinen impliziert daher teilweise oder globale Entfaltung, chemischen Austausch und die Rückfaltung in den nativen Zustand nach Gleichung (1) und die beobachteten Wechselkurse k obs abhängig von der Öffnungsrate k op, die Schlussrate k cl und der intrinsischen chemischen Austausch rate ch k gemäß Gleichung (2).
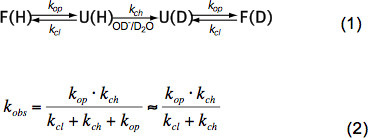
Unter nativen Zustand Bedingungen k op ist viel kleiner als k ch und kann im Nenner vernachlässigt werden. Es gibt zwei extreme Wechsel Regime genannt EX1 und EX2. Wenn der k Cl ist viel kleiner als k CH (EX1) die beobachtete Geschwindigkeit praktisch gleich der Öffnungsrate und HX unmittelbare Beobachtung der Entfaltung eines Strukturelements. Ein solcher Austausch Regime, in dem alle Amidprotonen Austausch auf einmal beim Öffnen des Strukturelements ist leicht beobachtbaren MS durch eine bimodale Verteilung der Isotopenpeaks 7. Wenn k Cl ist viel größer als k CH (EX2) die beobachtete Rate ist proportional zu k ch wobei die Proportionalitätskonstante gleich dem Faltungs-Entfaltungs-Gleichgewichte ist die Konstante K u = K op </sub> K / cl. Unter diesen Bedingungen sind viele Öffnungs-und Schließereignisse notwendig, bevor alle Amidprotonen gegen Deuteronen, was zu einem allmählichen Anstieg der Durchschnittsmasse, während die Isotopenverteilung etwa gleich bleibt. Die EX2 Regime ermöglicht die Bestimmung der freien Energie der Entfaltung &Dgr; G u und damit der Stabilität eines Strukturelements. Unter nativen Zustand Zustand der EX2-Regime ist am häufigsten. Erhöhung des pH-Wertes und Zugabe von chaotropen Mitteln können die Austauschmechanismus zu EX1 verschieben. Daher kann HX-MS verwendet werden, um thermodynamische erkunden sowie kinetischen Parameter der Proteinfaltung und Konformationsänderungen werden.
Wie oben erwähnt ist HX eigen pH-und temperaturabhängig und der Austausch Halbwertszeit von einer völlig löse ausgesetzt Protonen des Rückgrats Amidgruppe zwischen 5-400 ms bei einem physiologischen pH-Wert (pH 7,6) und 30 ° C, aber mindestens 10 min auf> 15 Stunden mit einem Mittelwert von> 2 h bei pH 2,9 und 0 °C (mit Ausnahme des Protons des ersten Gerüst-Amidbindung eines Polypeptids, das mit einer Halbwertszeit von ca. 1-2 min austauscht). Unter solchen langsamen Austausch Bedingungen ist es möglich, die Probe zu verdauen Proteasen (z. B. Pepsin), die aktiv sind, unter diesen Bedingungen mit verlieren sich alle in den eingeDeuteRonen enthaltenen Informationen. Seit der Einführung des Magen-Darm-Verdauung unter langsamem Austausch Bedingungen kann nicht nur die Gesamt HX Kinetik der vollständigen Proteine analysiert werden, aber HX kann auf bestimmte Regionen 8,9 lokalisiert werden. Ortsauflösung wird noch auf die Größe der Fragmente Magen erzeugt, die im allgemeinen zwischen 10-30 Reste beschränkt. Jedoch könnte überlappende Fragmente aufgrund der unspezifischen Art der Spaltung durch Pepsin erzeugt eine Erhöhung der räumlichen Auflösung führen. Darüber hinaus wurden mehrere andere Proteasen gefunden unter Abschrecken Bedingungen aktiv zu sein, aber viel weniger effizient als Pepsin 10. Weitere zunehse der räumlichen Auflösung können durch Fragmentierung von Peptiden in der Gasphase durch Methoden, die Deuterierung Muster wie Elektroneneinfang-Dissoziation (ECD), Elektronentransfer-Dissoziation (ETD) und Infrarot-Multiphoton-Dissoziation (IRMPD) 11-13 erhaltenen erreichen. Diese Methoden verhindern den Verlust an räumlicher Auflösung durch intramolekulare Protonenwanderung ("Scrambling"), die durch stoßinduzierte Dissoziation (CID) zu beobachten ist die am häufigsten verwendeten Fragmentierungstechnik. Allerdings erfordern diese Verfahren die Optimierung für jedes einzelne Peptid und ist damit immer noch ziemlich schwierig.
HX-MS wurde verwendet, um Protein-Liganden und Protein-Protein-Wechselwirkungen, einschließlich virale Kapsid-Zusammensetzung 14-17 analysieren. Protein-Entfaltung und Rückfaltung sowie temperaturinduzierte Konformationsänderung untersucht 7,18,19. Phosphorylierung und einzigen Aminosäure mutationsbedingte Konformationsänderungen 16,20 und nucleotide-induzierten Veränderungen wurden analysiert 21,22. Daher scheint diese Methode ideal geeignet zur Montage und Dynamik von molekularen Maschinen zu analysieren. Ein attraktiver Kandidat, dessen Mechanismus ist von großem allgemeinem Interesse ist das Hsp90-Chaperon-Komplex.
