Il numero di strutture cristalline di proteine e complessi proteici aumentato rapidamente negli ultimi anni. Essi presentano inestimabili istantanee della organizzazione strutturale di queste proteine e forniscono una base per l'analisi struttura-funzione. Tuttavia, la dinamica delle proteine e dei cambiamenti conformazionali, che sono essenziali per le loro funzioni, sono raramente rivelate da cristallografia a raggi X. Crio-electronmicroscopy, invece, è in grado di catturare proteina e proteina complessi in differenti conformazioni ma generalmente non possono risolvere cambiamenti conformazionali fino a livello di struttura secondaria 1. Dinamica conformazionale delle proteine in soluzione a dettagli atomiche possono essere risolti solo da NMR, ma questo metodo è ancora limitato alle proteine di relativamente piccole dimensioni (generalmente ≤ 30 kDa) e ha bisogno di alte concentrazioni di proteine (≥ 100 micron), che ostacola gli esperimenti con oligomerizzazione o aggregazione di proteine incline 2. Un metodo cheè in grado di colmare tra l'alta risoluzione cristallografia a raggi X e crio-electronmicroscopy e che non è limitata dalla dimensione proteine o concentrazione è ammide idrogeno-1 H / 2 H-scambio (HX) in combinazione con spettrometria di massa (MS). Negli ultimi anni questo metodo è sviluppata per uno strumento analitico che per l'analisi della dinamica delle proteine, folding, la stabilità della proteina e cambiamenti conformazionali 3-5. La base molecolare di questo metodo è la natura labile di backbone idrogeni ammidici in proteine, che si scambieranno con atomi di deuterio quando la proteina viene posto in un D 2 O soluzione. Il conseguente aumento della massa proteica nel tempo viene misurata con alta risoluzione MS.
In brevi peptidi strutturati HX dipende solo dalla temperatura, concentrazione di catalizzatore (OH -, H 3 O + pH, vedi Figura 3) e catene laterali di aminoacidi residui adiacenti a causa induttiva, gattoeffetti alytic e sterici. Questi effetti sul intrinseca scambio chimico tasso k ch sono state elegantemente quantificata Bai et al. 6 e un programma è disponibile (cortesia Z. Zhang), che calcola k ch per ogni amminoacido in un polipeptide dipende dal pH e temperatura. A pH neutro e temperature ambiente k ch è dell'ordine di 10 1 -10 3 sec -1. Nelle proteine ripiegate HX può essere 2-9 ordini di grandezza più lento dovuto principalmente legame idrogeno nella struttura secondaria e in misura minore a causa di accesso ristretto di idrati ioni OH – all'interno di una proteina strettamente ripiegata. HX in proteine native implica quindi dispiegarsi, a scambio chimico parziale o globale e ripiegamento allo stato nativo secondo l'equazione (1) e ai tassi di cambio osservati k obs dipendono op tasso di apertura k, il tasso di chiusura k cl e lo scambio chimico intrinseco raTE k ch secondo l'equazione (2).
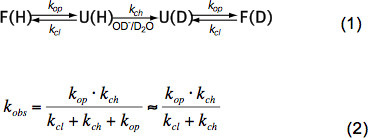
In condizioni di stato nativo k op è molto più piccolo di k ch e può essere trascurato nel denominatore. Ci sono due regimi di cambio estreme chiamati EX1 ed EX2. Se k cl è molto più piccolo k ch (EX1) il tasso osservato è praticamente uguale al tasso di apertura e HX permette l'osservazione immediato dello svolgimento di un elemento strutturale. Un tale regime di scambio, dove ogni scambio contemporaneamente protoni ammidici all'apertura dell'elemento strutturale, è facilmente osservabile in MS da una distribuzione bimodale dei picchi isotopici 7. Se k cl è molto maggiore di k ch (EX2) il tasso osservato è proporzionale k ch cui la costante di proporzionalità è uguale equilibri-piegatori dispiegarsi costante K u = k op </sub> / K cl. In queste condizioni, molti eventi di apertura e chiusura sono necessarie prima di effettuare cambi protoni ammidici per deuteroni, portando ad un graduale aumento della massa media mentre la distribuzione isotopica rimane sostanzialmente identico. Il regime EX2 consente la determinazione dell'energia libera di dispiegarsi ΔG u e quindi la stabilità di un elemento strutturale. In condizioni stato nativo il regime EX2 è più comune. Aumento del pH e aggiunta di agenti caotropici può spostare il meccanismo di scambio di EX1. Pertanto, HX-MS può essere utilizzato per esplorare termodinamico e parametri cinetici di folding e cambiamenti conformazionali.
Come accennato in precedenza HX è intrinsecamente pH e temperatura dipendente e lo scambio emivita di un protone completamente solvente esposta del gruppo ammidico backbone è tra 5-400 msec a pH fisiologico (pH 7,6) e 30 ° C, ma 10 min a> 15 ore con una media di> 2 ore a pH 2,9 e 0 °C (tranne per il protone del primo backbone ammide legame di un polipeptide, che scambia con un'emivita di ca. 1-2 min). In tali condizioni lento scambio è possibile digerire l'esempio utilizzando proteasi (ad esempio pepsina) che sono attivi in queste condizioni, con fuori perdere tutte le informazioni contenute nei deuteroni incorporati. Dopo l'introduzione di digestione peptica in condizioni scambio lento, non solo la cinetica complessiva HX di proteine integrali possono essere analizzati ma HX possono essere localizzati alle regioni specifiche 8,9. Risoluzione spaziale è attualmente limitata alle dimensioni dei frammenti generati peptiche, che è in generale tra 10-30 residui. Tuttavia, frammenti sovrapposti creati a causa della natura non specifica di clivaggio con la pepsina potrebbe portare ad un aumento della risoluzione spaziale. Inoltre, diverse altre proteasi sono risultati essere attiva in condizioni di tempra, tuttavia, molto meno efficiente rispetto pepsina 10. Ulteriore Increase di risoluzione spaziale può essere raggiunto con la frammentazione dei peptidi in fase gassosa con metodi che conservano il modello deuterazione come cattura di elettroni dissociazione (ECD), il trasferimento di elettroni dissociazione (ETD) e infrarossi multiphoton dissociazione (IRMPD) 11-13. Queste tecniche prevenire la perdita di risoluzione spaziale dovuta alla migrazione intramolecolare protone ("scrambling"), che si osserva per dissociazione indotta da collisione (CID) la frammentazione tecnica più comunemente usata. Tuttavia, questi metodi richiedono l'ottimizzazione per ogni singolo peptide ed è quindi ancora molto impegnativo.
HX-MS è stato utilizzato per analizzare proteina-ligando e proteina-proteina interazioni compresi l'assemblaggio del capside virale 14-17. Proteine dispiegamento e ripiegamento, nonché di temperatura indotti cambiamenti conformazionali sono stati studiati 7,18,19. Fosforilazione e singolo aminoacido mutazione legata conformazionale cambiamenti 16,20 e nucleotcambiamenti ide-indotti sono stati analizzati 21,22. Pertanto, questo metodo sembra idealmente adatto per analizzare il montaggio e dinamica di macchine molecolari. Un candidato attraente, il cui meccanismo è di grande interesse generale, è il complesso chaperone Hsp90.
