Количество кристаллических структур белков и белковых комплексов резко увеличилось в последние годы. Они представляют бесценные снимки структурной организации этих белков и обеспечить основу для структуры и функции анализа. Тем не менее, динамика белков и конформационных изменений, которые необходимы для их функций, которые редко показал с помощью рентгеновской кристаллографии. Крио-Электронная микроскопия, с другой стороны, имеет возможность захвата белка и белковых комплексов в различных конформаций, но в целом не может решить конформационные изменения до среднего уровня структуры 1. Конформационная динамика белков в растворе на атомном деталей может быть решен только с помощью ЯМР, но этот метод по-прежнему ограничен белков относительно небольших размеров (обычно ≤ 30 кДа) и нуждается в высокой концентрации белков (≥ 100 мкм), что затрудняет эксперименты с олигомеризации или агрегации, склонные белков 2. Одним из методов,способна преодолеть между высоким разрешением рентгеновской кристаллографии и крио-Электронная микроскопия и который не ограничен размером белка или концентрации водорода является амид-1Н / 2 Н-обменного (HX) в сочетании с масс-спектрометрией (MS). В последние годы этот метод разработан для ценного аналитического инструмента для анализа динамики белков, сворачивания белка, стабильности белка и конформационных изменений 3-5. Молекулярные основы этого метода является лабильным характер системообразующих амидных атомов водорода в белках, которые будут обмениваться с атомами дейтерия, когда белок помещается в D 2 O раствора. Последующее увеличение массы белка с течением времени измеряется с высоким разрешением MS.
В коротких неструктурированных пептидов HX зависит только от температуры, концентрации катализатора (ОН -, Н 3 О + т.е. рН, см. рисунок 3) и боковых цепей аминокислот из соседних остатков из-за индуктивной, кошкиalytic и пространственные эффекты. Эти эффекты на внутренней химической обменный курс К ч были элегантно количественно Бай и др.. 6 и программа доступна (любезность З. Чжан), который вычисляет К ч для каждой аминокислоты в полипептид в зависимости от рН и температуры. При нейтральном рН и температуре окружающей среды К ч составляет порядка 10 1 -10 3 с -1. В свернутых белков HX может быть 2-9 порядков медленнее в основном за счет водородных связей в вторичной структуры и в меньшей степени за счет ограниченного доступа гидратированных ОН – ионов к внутренней части плотно уложенного белка. HX в нативных белков поэтому подразумевает частичную или глобальной разворачивается, химический обмен и повторной укладки в нативном состоянии в соответствии с уравнением (1) и наблюдаемые курсы обмена K набл зависеть от темпов открытия К ор, скорость закрытия К кл и внутренней химического обмена раТе К ч в соответствии с уравнением (2).
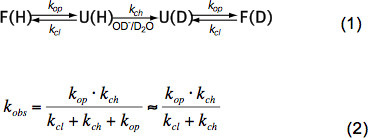
Под носителями государственных условиях к оп намного меньше, чем К ч и ими можно пренебречь в знаменателе. Есть два крайних режимов обмена называемые EX1 и EX2. Если к кл намного меньше, чем К ч (EX1) наблюдаемая скорость практически равна скорости открытия и HX позволяет сразу же наблюдение разворачивание структурного элемента. Такой обмен режим, где все амидные протоны обмен сразу при открытии структурного элемента, легко наблюдаемая в MS бимодальным распределением изотопов пиков 7. Если к кл намного больше, чем К ч (EX2) наблюдаемая скорость пропорциональна К ч причем коэффициент пропорциональности равен складных-разворачивания равновесий постоянная К U = K оп </sub> К / кл. В этих условиях многие открытия и закрытия мероприятия необходимы, прежде всего обмен амидные протонов для дейтронов, что приводит к постепенному увеличению средней массы в то время как изотопный распределение остается примерно такой же. Режим EX2 позволяет определить свободной энергии разворачивается ΔG и и поэтому стабильность структурного элемента. Под родной государственной состоянии режим EX2 является наиболее распространенным. Повышение рН и добавлением хаотропных агентов можно сдвинуть обменный механизм EX1. Таким образом, HX-MS может быть использован для изучения термодинамических а также кинетические параметры складывания белка и конформационных изменений.
Как упоминалось выше HX внутренне рН и температура зависят и обмен полураспада полностью открытой растворителя протона магистральной амидной группы находится между 5-400 мс при физиологическом значении рН (рН 7,6) и 30 ° С, но 10 мин до> 15 час при среднем значении> 2 ч при рН 2,9 и 0 °С (для протона первого магистральной амидной связи полипептида, который обменивается с периодом полураспада ок. 1-2 мин исключением). В таких медленно обменивающихся условиях можно переварить примера с использованием протеазы (например пепсин), которые активны в этих условиях, с нашими потерять всю информацию, содержащуюся в объединенных дейтронов. С момента введения язвенной пищеварения при медленных условиях обмена, не только общие HX кинетика полноразмерные белки могут быть проанализированы, но HX могут быть локализованы в конкретных регионах 8,9. Пространственное разрешение в настоящее время ограничивается размером пептической фрагментов, полученных, которая является в целом 10-30 остатков. Тем не менее, перекрывающиеся фрагменты, созданные за счет неспецифического характера расщепления пепсином может привести к увеличению пространственного разрешения. Кроме того, несколько других протеаз, оказались активными при условиях закалки, однако, значительно менее эффективен, чем пепсина 10. Кроме того увеличилисьсебе пространственного разрешения может быть достигнуто путем фрагментации пептидов в газовой фазе методами, которые сохранились дейтерирование шаблон например захвата электронов диссоциации (ECD), перенос электрона диссоциации (ETD) и инфракрасного многофотонная диссоциация (ИКМФД) 11-13. Эти методы предотвращения потери пространственного разрешения в связи с внутримолекулярной миграции протонов ("скремблирования"), которое наблюдается на столкновительное диссоциации (CID) наиболее часто используется методика фрагментации. Однако эти методы требуют оптимизации для каждого отдельного пептида и, таким образом, все еще довольно сложной задачей.
HX-MS была использована для анализа белок-лиганд и белок-белковых взаимодействий в том числе вирусного капсида сборки 14-17. Белок разворачивается и повторной укладки, а также температуры, вызванные конформационные изменения были исследованы 7,18,19. Фосфорилирования и один аминокислотный мутация связанных конформационные изменения 16,20 и nucleotязь-индуцированные изменения были проанализированы 21,22. Таким образом, этот метод кажется идеально подходит для анализа сборки и динамики молекулярных машин. Один привлекательным кандидатом, механизм которого имеет большое общий интерес, является компаньонка комплекс Hsp90.
