Figure 2 shows representative polyribosome profiles after fractionation. The profiles of brain and spinal cord show characteristic absorption curves as described before for cell lines. mRNAs bound to small ribosomal subunits (40S) sediment at lighter fractions and appear first as a peak on the profile, followed by the large ribosomal subunit (60S) and monosome (80S) bound mRNAs. mRNAs bound to multiple ribosomes sediment at heavier fractions, with the later peaks indicating increasing number of bound ribosomes. Global translation can be assessed from the profile. As an example, brain tissue has a higher polyribosome to monosome ratio than spinal cord tissue, indicating more active translation.
RNA extracted from individual fractions were assessed by Bioanalyzer, showing distribution of 18S and 28S rRNA. 18S rRNA appears earlier in the profile, in accordance with the small ribosomal subunits sedimenting in lighter sucrose fractions. Typical yields of total RNA are 10-20 µg for brain and 2-4 µg for spinal cord. RNA yields from individual fractions are up to 4 µg and 0.8 µg for brain and spinal cord respectively, depending on the fraction. A blank sucrose gradient already shows some background absorption at 260 nm, due to the presence of DTT. This background can be subtracted during data analysis in order to normalize sample values. EDTA treatment collapsed the polyribosome peaks, demonstrating the sedimentation profile is due to translation.

Figure 1. Workflow and potential applications of in vivo polyribosome fractionation. Please click here to view a larger version of this figure.
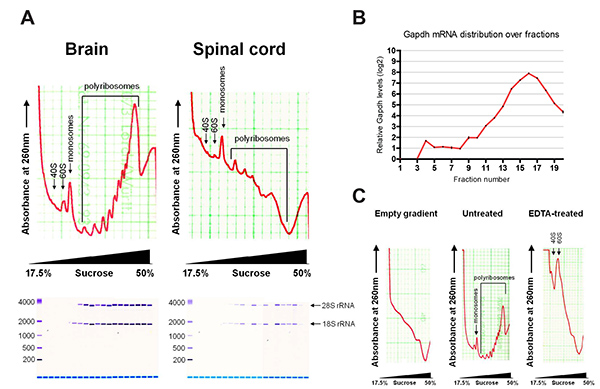
Figure 2. (A) Typical polyribosome profiles of brain and spinal cord, with assessment of extracted RNA by Bioanalyzer. (B) Distribution of Gapdh mRNA over fractions by qRT-PCR. (C) Polyribosome profiles of empty gradient and brain with and without EDTA treatment. Please click here to view a larger version of this figure.
| 1.1 | 2x gradient buffer | 30 mM Tris-HCl pH7.4, 30 mM MgCl2, 600 mM NaCl, 200 µg/ml cycloheximide (CHX), 2mM dithiothreitol (DTT) | |
| 1.1 | sucrose solution 17.5% | 8.67 g sucrose, 25 ml 2x gradient buffer, up to 50 ml DEPC H20 | |
| 1.1 | sucrose solution 25.6% | 12.8 g sucrose, 25 ml 2x gradient buffer, up to 50 ml DEPC H20 | |
| 1.1 | sucrose solution 33.8% | 16.9 g sucrose, 25 ml 2x gradient buffer, up to 50 ml DEPC H20 | |
| 1.1 | sucrose solution 41.9% | 20.95 g sucrose, 25 ml 2x gradient buffer, up to 50 ml DEPC H20 | |
| 1.1 | sucrose solution 50% | 25 g sucrose, 25 ml 2x gradient buffer, up to 50 ml DEPC H20 | |
| 2.3 | homogenization buffer A | 0.25 M sucrose, 50 mM Tris/HCl, pH7.4), 5 mM MgCl2, 25 mM KCl | 200 µg/ml CHX, 1x Roche complete protease inhibitor, 1 mM DTT, 1mM phenylmethanesulfonylfluoride (PMSF), 100 U/ml RNAsin |
| 3.1 | 2M sucrose solution | 68.4% sucrose, 50 mM Tris-HCl, pH7.4, 5 mM MgCl2, 25 mM KCl | 100 µg/ml CHX, 1x Roche complete protease inhibitor, 1 mM DTT, 1 mM PMSF |
| 3.1 | 1.1M sucrose solution | 38.5% sucrose, 50 mM Tris-HCl, pH7.4, 5 mM MgCl2, 25 mM KCl | 100 µg/ml CHX, 1x Roche complete protease inhibitor, 1 mM DTT, 1 mM PMSF |
| 3.1 | 0.9M sucrose solution | 30.8% sucrose, 50 mM Tris-HCl, pH7.4, 5 mM MgCl2, 25 mM KCl | 100 µg/ml CHX, 1x Roche complete protease inhibitor, 1 mM DTT, 1 mM PMSF |
| 4.1 | homogenization buffer B | 0.25 M sucrose, 50 mM Tris/HCl, pH7.4, 5 mM MgCl2, 25mM KCl, 1% NP-40, 1% sodium deoxycholate | 200 µg/ml CHX, 1x Roche complete protease inhibitor, 1mM DTT, 1 mM PMSF , 100 U/ml RNAsin |
Table 1. List of buffers and solutions.
