Here, we describe a representative heart dissection experiment using the zebrafish Tg(myl7:GFP)twu34 transgenic line9, which expresses green fluorescent protein (GFP) exclusively within the myocardium (Fig. 1). We collected both the dissected hearts and the embryos from which they were derived to assess the purity of the heart sample. Briefly, homozygous Tg(myl7:GFP)twu34 zebrafish9 were outcrossed with wild-type so that all embryonic hearts were GFP labeled. Some 500 embryos (56 hpf) were transferred into 1.5 ml tubes (approx. 100 embryos in each) (Fig. 1A1, 1B). Each tube was processed as described in the Protocol section (step 2) and in Figure 1A. Hearts were released by pipetting up and down several times (Fig. 1A2) and then applied onto a 100 μm filter (Fig. 1A3). Large pieces of embryonic tissue were retained in this filter; this fraction was retrieved and put in Trizol for RNA extraction to obtain an “embryo without heart” RNA sample (Fig. 1A3). The flow-through containing hearts was then applied onto a 30 μm filter (Fig. 1A4), which retained the hearts and tissue debris of similar size. GFP-positive hearts were flushed out of the filter into an agarose-coated petri dish (Fig. 1A5, 1C), quickly gathered in the middle of the dish under a fluorescent stereomicroscope and transferred into 0.75 ml RNAlater (Fig. 1A6). Within this tube with RNAlater, we pooled hearts derived from 5 dissection rounds (approx. 300 hearts from 500 embryos representing an extraction efficiency of about 60%). A comparison of the heart samples prior to sorting from the debris from embryos at 56 hpf (Fig. 1C) versus 36 hpf (Fig. 1C’) is shown in Figure 1. At 56 hpf, the disruption of embryos yielded more embryonic debris (such as lenses, arrowhead, Fig. 1C) than at 36 hpf following the 30 μm filtration step (Fig. 1C’).
To determine the purity of the 56 hpf “heart” sample, we compared the expression levels of cardiac versus extra-cardiac transcripts in “heart” and “embryo without heart” samples by RT-qPCR. To this end, we extracted mRNA from those two samples and assessed RNA quality on an agarose gel (Figure 2A) and quantity by absorbance measurement (Table 1) as described in the protocol section (step 3). The approx. 300 hearts yielded 660 ng RNA. Next, cDNA was synthesized using M-MLV Reverse transcriptase RNase H(-) and random hexamers according to Manufacturer´s instructions, and RT-qPCR experiments were performed as described12. Relative gene expression levels were calculated for different tissue-specific markers such as myosin light polypeptide 7 [myl7, a myocardial marker]9, kinase insert domain receptor like [kdrl, an endothelial/endocardial marker]13, hemoglobin beta embryonic-1.1 [hbbe1.1, an erythrocyte marker]14, crystallin-alpha A [cryaa, an eye lens marker]15, and glial fibrillary acidic protein [gfap, a central nervous system marker]16 (Figure 2B). The zebrafish eukaryotic translation initiation factor 1B [eif1b]12 was used as an internal reference gene (see Table 2 for GenBank ID numbers, primer sequences, PCR product sizes and tissue specificity for each gene). As expected, myl7 was highly enriched in the “heart” sample as compared to the “embryo without heart” sample (Fig. 2B). The endothelial cell marker kdrl was found to be moderately enriched in the heart sample, as expected (about 40% of the cardiac cells at 56 hpf are kdrl-expressing endocardial cells) (Fig. 2B). The erythrocyte marker hbbe1.1 was overrepresented in the “heart” sample, even though the hearts were still beating after dissection, which should expel red blood cells from the heart (Fig. 2B). In contrast, the expression levels of the CNS and lens markers (gfap and cryaa, respectively) were extremely low in the “heart” sample (Fig. 2B). Altogether, we conclude that the heart dissection protocol yields high quality and cardiac-specific RNA from entire zebrafish embryos.
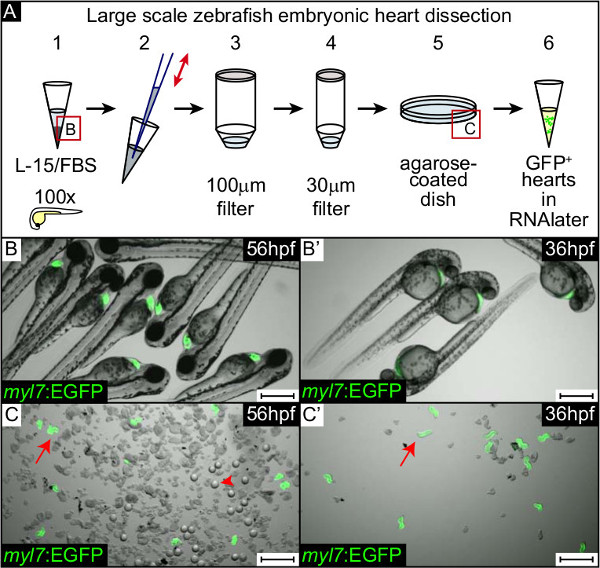
Figure 1. Heart extraction from zebrafish embryos. (A) Scheme depicting the heart extraction procedure. About 100 live embryos are collected in a 1.5 ml tube (A1, B,B’), anesthetized and then pipetted up and down several times through a narrow pipette tip to release the hearts (A2). The resulting solution containing the embryonic tissues (A2) is then applied onto a 100 μm filter (A3). Embryonic tissues without yolk and hearts, and large embryonic debris remain in the filter (A3). The flow-through is collected in a 50 ml tube (A3) and then applied onto a 30 μm filter (A4). The hearts and small debris are retained in the 30 μm filter and transferred into a small agarose-coated petri dish (A5, C,C’). EGFP labelled hearts (C,C’; arrow) are manually sorted from the remaining debris, such as lenses (C, arrowhead), and are collected in RNAlater (A6). This procedure is repeated at least 5 times and all hearts are pooled together for further processing. (B-C’) Overlay of fluorescence (EGFP in green) and DIC images of live transgenic Tg(myl7:EGFP)twu34 embryos at 56 hpf (B,C) and 36 hpf (B’,C’) at two steps of the heart dissection procedure: prior to embryo dissection (B,B’) and just after flushing from the 30 μm filter, prior to sorting the hearts from the debris in the petri dish (C,C’). Scale bar: 500 μm.
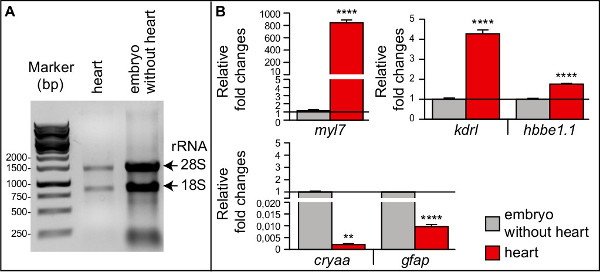
Figure 2. Analysis of RNA quality and purity by electrophoresis and RT-qPCR, respectively. (A) Total RNA from the “heart” sample (2 μl) and from the “embryo without heart” sample (1 μl), derived from 56 hpf embryos, resolved on a 1% agarose gel. Prominent S18 and S28 rRNA bands indicate that RNAs are not degraded. (B) Relative expression levels of the tissue specific markers myl7 (myocardium), kdrl (endothelium), hbbe1.1 (erythrocytes), cryaa (lens), and gfap (CNS) as determined by RT-qPCR for the “heart” sample normalized to the “embryo without heart” sample. The RT-qPCR experiments were performed as technical triplicates and data are given as means ± SEM. An unpaired t-test analysis was performed. **** p<0,0001; ** p<0,005. bp, base pair.
| sample ID | ng/μl | A260 | A280 | 260 / 280 | 260 / 230 | cursor abs. | 340 raw |
| hearts | 32.97 | 0.82 | 0.41 | 2.01 | 0.35 | 2.355 | 0.031 |
| embryos without hearts | 568.82 | 14.22 | 7.00 | 2.03 | 2.04 | 6.965 | 0.018 |
Table 1. RNA quantity and quality by spectrophotometric measurement. The quantity and quality of the RNA of the “heart” and “embryo without heart” samples obtained from 56 hpf embryos as measured by spectrophotometry.
| gene | GenBank # | primer sequence | PCR product size (bp) | Tissue-specific marker |
| myl7 | BX248505 | F: 5'-ACAGCAAAGCAGACAGTGAA-3' | 163 | myocardium |
| R: 5'-TAACTCCATCCCGGTTCTGA-3' | ||||
| kdrl | CR759732 | F: 5'-ACAACGACACTGGCATCTAC-3' | 170 | endothelium/ endocardium |
| R: 5'-TGTTCTACAGGGGACCACAA-3' | ||||
| gfap | BX324157 | F: 5'-AGACAACTTGGCCTCAGAC-3' | 247 | central nervous system |
| R: 5'-ATCCACATGAACCTGTTGGG-3' | ||||
| cryaa | BX248514 | F: 5'-ATCCAACACCCTTGGTTCAG-3' | 190 | eye lens |
| R: 5'-TCAGACCTCACCTCAGAGAC-3' | ||||
| hbbe1.1 | BC095024 | F: 5'-TGGTTGTGTGGACAGACTTCGA-3' 8 | 102 | erythrocyte |
| R: 5'-CGATAAGACACCTTGCCAGAGC-3' 8 | ||||
| eif1b | BX323079 | F: 5'-CAGAACCTCCAGTCCTTTGATC-3' 12 | 195 | reference gene |
| R: 5'-GCAGGCAAATTTCTTTTTGAAGGC-3' 12 |
Table 2. Primers used for the RT-qPCR experiments. Name, GenBank accession number, sequence, PCR product sizes and tissue-specificity are shown for all genes analyzed.
