Una comprensión más fundamental de cómo las células T reconocen los antígenos requiere que mira en el lugar correcto, es decir, dentro de la sinapsis inmunológica formada entre la célula T y el APC. Aquí, la cinética de unión molecular no sólo están determinadas por las propiedades bioquímicas inherentes de los compañeros de interacción involucrados, pero dependerá en gran medida de parámetros celulares, que incluyen fuerzas celulares, la arquitectura de la membrana y las interacciones laterales entre proteínas de la membrana así como las limitaciones geométricas-sinapsis específica 3. Enfoques bioquímicos están limitados en poder de resolución, ya que requieren la interrupción de al menos una de las membranas sinápticas implicadas. Por este motivo se ha desarrollado una metodología de formación de imágenes basada en FRET para monitorear la unión del TCR a pMHCs antigénicos 2. Aquí las células T están decoradas con un recombinante y el sitio específicamente etiquetados fragmento de anticuerpo de cadena simple-TCRβ reactiva (SCF V) y se enfrentaron con el plan bicapas ar apoyado por el vidrio de lípidos (SLBS), que albergan MHC moléculas clase II cargadas con un péptido antigénico marcado con fluorescencia, moléculas coestimuladoras y proteínas de adhesión. Unión sináptica entre marcado tinte TCR y marcado con tinte resultados pMHC en FRET, que pueden ser monitoreados en un mayor y el nivel de moléculas individuales por Total Interna Fluorescencia de Reflexión (TIRF) microscopía.
En este artículo se explica en detalle cómo utilizar SLBS para ensayar las sinapsis de células T, verificar su integridad a través de un ensayo de flujo de calcio de células T funcional, conducta FRET mediciones a granel y con sensibilidad única molécula, y analizar los datos adquiridos. Las recomendaciones se ofrecen para la producción de proteínas adecuadamente conformados necesarios para funcionalización bicapa. Para obtener información más específica acerca de la formación de bicapa y la configuración de un microscopio TIRF adecuada consulte a un acceso público JoVe publicación adicionales publicados espalda con espalda 4.
tienda "> Naturaleza de SLBSSLBS funcionalizables se pueden generar fácilmente a partir de vesículas unilamelares (SUV) que contienen los dos lípidos-2-oleoil-sn-gylcero-3-fosfocolina 1-palmitoil (corto: POPC, 90-99%) y sn-1,2 dioleoyl- – glicero-3 – {[N (5-amino-1-carboxipentil) iminodiacético ácido] succinil} (corto: DGS NTA-Ni, 10.01%). Todo terrenos repartidos en portaobjetos de vidrio limpios para formar una bicapa plana contigua 4. DGS-NTA-Ni sirve para anclar proteínas marcada con polihistidina a través de la formación de complejo de polihistidina mediada con el NTA-Ni-que contiene grupo de cabeza (Figura 1A) sintético. Por asociación estable de un típicamente sustituye al dominio transmembrana nativa y cola citoplásmica de la proteína de adhesión ICAM-1 y la molécula coestimuladora B7-1 con una etiqueta que contiene doce histidinas (ICAM-1-12H, -12H B7-1) (Figura 1B) . La clase II molécula IE k cargado con péptido contiene dos cadena polipeptídica membrana incorporado (α y β)s. Los transmembrana / dominios citoplásmicos de ambas cadenas tienen que ser reemplazados con una etiqueta que contiene seis histidinas cada uno (IE k α β 6H 6H o IE k -2x6H). Como alternativa, la ampliación de la α-cadena con doce histidinas y dejando el dominio extracelular de la cadena β sin etiquetar (dando lugar a IE k α β 12H 0H o IE k -12H) da lugar a resultados satisfactorios (Figura 1B).
Etiquetado específico del sitio de pMHCs
Es importante para etiquetar el pMHC estequiométricamente y específica de sitio con el fin de ser capaz de convertir los rendimientos medidos FRET en las constantes de unión de equilibrio significativas. Esto se puede lograr por medio del etiquetado química de un péptido sintético que se carga en la hendidura de unión al péptido de histidina recombinante marcada con moléculas MHC de clase II 2,5. El péptido incluye todos los residuos del epítopo de células T como well como un corto C-terminal enlazador (GGS) seguido de cisteína (por ejemplo, en el citocromo c de polilla (MCC) péptido ANERADLIAYLKQATK- GGSC, el enlazador está marcado en negrita). Esta cisteína se utiliza para marcar el péptido estequiométricamente con el uso de derivados de maleimida en colorantes. En este punto, un cuidado especial debe ser dedicado a la verificación cuantitativa tinte acoplamiento al péptido que contiene cisteína. Se recomienda HPLC-purificación del producto de adición de péptido-colorante y tiene que ser seguido por espectroscopia de masas de ionización por electrospray. Cualquier masas grabados correspondientes al educto péptido (sin colorantes) reflejan etiquetado incompleto. Si esto es cierto, el péptido purificado por HPLC debe ser sometido a rondas consecutivas de tinte etiquetado hasta el etiquetado se considera cuantitativo. Tenga en cuenta que la espectroscopia de masas MALDI-TOF se debe evitar ya que este método implica radiación láser para la muestra de ionización. Este tratamiento se desintegra los fluoróforos sensibles conectados antes de que se dio lectura a la masa péptido y así underrepre senta grados de tinte de conjugación.
El marcaje indirecto aún específica de sitio de los TCR unidos a células con el uso de una sola cadena monovalente fragmentos F V
Todavía es un reto para fijar los tintes a teléfonos proteínas asociadas a la superficie de las células vivas de una manera específica de sitio. Para superar este obstáculo para los TCR expuestos en la superficie, una versión monovalente de cadena sencilla (scFv V) de los genes del anticuerpo monoclonal Reactivo TCRβ H57-197 2 se ha construido. La estructura cristalina de este anticuerpo en complejo con el TCR permite diseñar racionalmente una versión, en la que un residuo de serina en estrecha proximidad a la C-terminal del péptido asociado de MHC (donde está conectado el correspondiente colorante pareja FRET) se sustituye por una residuo de cisteína. Esta cisteína mutante sirve entonces como un aceptor para la conjugación colorante (Figura 2).
Metodologías para grabar FRET
nt "> valores de FRET a granel son los más adecuados para verificar la relación entre las distancias inter-dye elegidos y FRET eficiencias medidas en este TCR-pMHC sistema 2 vinculante. Además, las mediciones de FRET a granel revelan diferencias cualitativas y cuantitativas en sinápticas afinidades TCR-pMHC ( ver abajo y sección de protocolo 3.2). Diversos enfoques para cuantificar la eficiencia de FRET se han introducido en la literatura 6. En este artículo FRET se registra a través de(a) la recuperación de los donantes después del blanqueo aceptante, y por medio de
(b) sensibilizado FRET emisión aceptor.
El primer método (a) requiere el uso de un aceptor de FRET que pueden ser photobleached fácilmente, y un donante, que es bastante fotoestable. Además, es importante asegurarse de que el aceptor photobleached ya no es capaz de extinguir la fluorescencia del donador. Como el mismo canal de detección (donante) se utiliza para la cuantificación, sin factores de corrección unad no hay aberraciones cromáticas tienen que ser considerados, lo que hace que esta metodología simple y fiable. Sin embargo, las mediciones cuantitativas no se pueden repetir en el mismo lugar espécimen y los cambios en la FRET no se pueden grabar en el tiempo. Para evitar los efectos causados por la difusión molecular o la motilidad celular una etapa de blanqueo rápido debe estar dirigida para, lo que minimiza el tiempo que pasa entre la primera donante de FRET (antes del blanqueo aceptor) y el segundo de adquisición de imágenes donante de FRET (después de FRET aceptor de blanqueo). Es recomienda emplear una poderosa fuente de luz láser de la longitud de onda de excitación aceptor FRET con el fin de reducir al mínimo la iluminación y el blanqueo de veces.
Por el contrario, en el enfoque de la medición de la emisión sensibilizada FRET (b) el donante de FRET es excitado y la emisión del aceptor de FRET se observa en el canal aceptor de FRET. Los cambios en la señal de aceptor de FRET se pueden grabar con el tiempo pero de emisión del donador FRET en el canal aceptor desplazada al rojo (termed bleedthrough) y FRET aceptora cruzada de excitación a través de la excitación de los donantes tienen que ser determinados y restado del canal aceptor FRET registrada con exactitud. Para ello, los correspondientes FRET donante y aceptor de FRET imágenes tienen que estar alineados espacialmente.
La detección de una sola molécula (cm) de FRET eventos
Con el uso de láseres como fuente de excitación, una cámara sensible y TIRF microscopía de ruido atenuado la fluorescencia de los fluoróforos individuales se pueden rastrear fácilmente con el tiempo. Similar es cierto para la detección de eventos smFRET intermoleculares. Sin embargo, las complicaciones pueden ser causados por FRET bleedthrough donante y cross-excitación del aceptor FRET, y por lo tanto mucho cuidado tiene que ser tomado al ajustar las densidades fluoróforo en el experimento smFRET.
En el protocolo se proporciona a continuación (servicio de protocolo 4) el TCR fue elegido como FRET donantes en alta abundancia y pMHC como FRET aceptora con poca abundancia. Para atenuar FRET donante bleedthrough suficientemente, decorar el 10-30% de los TCR con fluorescente scFv V y 90 a 70% de los TCR con no fluorescente scFv V. Aquí el canal aceptor de FRET fue elegido como único canal molécula porque es confocal con la molécula de FRET solo canal. Esto ayuda a alinear eventos smFRET con aceptores de FRET sola molécula, que es la base de la validación smFRET.
La extracción fuera de las tasas a través de mediciones sinápticas smFRET
Photobleaching tanto FRET donante y aceptor FRET tiene que ser contabilizadas al extraer la vida media de las interacciones de una sola molécula FRET rastros. El número de FRET señales observables en el comienzo de su aparición como único par donador-aceptor N (0) se reduce con el tiempo por tanto unbinding del photobleaching complejo y receptor-ligando. El número de complejos de sobrevivir en un momento dado N (t) se puede expresar matemáticamente como sigue:
<p class = "jove_content">
En el término exp photobleaching (-t / τ lejía) el tiempo t es descrito por el producto del número de observaciones n y el tiempo de iluminación t enferma a causa del modo de observación no continua, discreta (es decir, solamente se produce la decoloración durante la iluminación ). Dentro de la exp- cinética plazo (t / τ off) el tiempo t es el producto del número de observaciones n y t el tiempo de retardo para una única observación FRET (es decir, unbinding cinética ocurre continuamente). Ecuación 1 se puede expresar como:
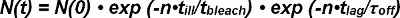
La τ término lejía / τ mal describes el número de observaciones hasta que se produzca el blanqueo y se define como el valor esperado <n blanquear> de su función exponencial. Ecuación 2 se puede simplificar como sigue:
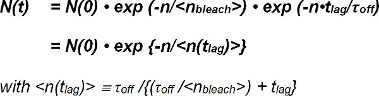
El valor esperado <n (t lag)> del número de cuadros N (t) con FRET-eventos observables después del tiempo t se determina directamente a partir del experimento. Depende del tiempo ajustable entre las observaciones (t lag) elegidos en el experimento y los valores desconocidos para τ off (el inverso de la tasa de descuento k off) y <n blanquear>, el valor esperado del número de observaciones antes de que ocurra el blanqueo .
Por lo tanto, el cálculo del valor esperado <n (t lag)> durante al menos dos valores de t <sub> lag permite la determinación experimental de <n blanquear> y T se fuera.
La extracción de los valores sinápticos 2D-K D a través de mediciones basado en FRET
La medición de la ocupación de un TCR, es decir, la relación entre la envolvente y las TCR TCR totales, es fundamental para determinar los valores sinápticos 2D-K D. Según la ecuación 4 este término es directamente proporcional a la medida FRET producir, siempre y cuando los TCR sirve como donantes y aceptores de FRET pMHCs como FRET.
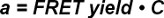
con una ocupación = TCR, C = factor de conversión
C es una constante, que depende del sistema de FRET y los fluoróforos utilizados. Se puede determinar experimentalmente como se muestra a continuación. una se puede convertir en un 2D-K D según la ecuación 5 cuando elSe sabe densidad inicial de ligandos de TCR antes de la adición de las células T a la bicapa. Esto es debido a la gran movilidad de las proteínas unidas-SLB y también porque SLBS proporcionar un depósito casi inagotable de ligandos 2.
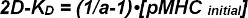
con [pMHC inicial] = densidad inicial de pMHC antes de la adición de las células T
Con las ecuaciones 4 y 5 ahora se puede determinar fácilmente la sináptica 2D-K D entre TCR y pMHC. Esto se hace más fiable con mediciones de FRET basado en la recuperación de los donantes después del blanqueo aceptor (ver sección 3.1 del protocolo).
Sin embargo, para medir C la relación entre la intensidad I FRET FRET (corregida para el fondo, FRET bleedthrough donante y aceptor de FRET transversal de excitación) y TCR ocupación A tiene que ser determinada. Para estetiene que saber la relación R entre la intensidad media de fluorescencia de los fluoróforos individuales asociados-TCR FRET donantes (por ejemplo Cy3 o AF555) sm me traste donante y la intensidad media de una sola molécula FRET eventos sm me preocupe. R depende del sistema de FRET en cuestión, filtros de emisión y de la cámara utilizada para la detección de fluorescencia.
El TCR de ocupación una continuación, se puede determinar directamente según la ecuación 6.

con R = sm me traste donante / sm me traste
R se determinó como 1,45 para el sistema H57 scFv Cy3 / pMHC-Cy5 conduce a:
a = mayor que FRET / mayor que TCR-cy3 • 1.45
La relación entre la ocupación de TCR A y el rendimiento FRET se puede determinar mediante FRET donante recuperary después del blanqueo aceptor. Para ello ambos parámetros se representan uno contra el otro para un número microclusters TCR como se muestra en la Figura 4A .La pendiente de la ajuste lineal indica el factor de conversión C (partir de la ecuación 4).
Como se ha demostrado en la Figura 4A, C asciende para (a) el scFv H57 V – Cy3 / Cy5-pMHC sistema de FRET y (b) la configuración del sistema microscopio aplicado a 1.995. El TCR de ocupación a puede fácilmente deducirse de la siguiente manera:
Ocupación TCR a = FRET ceder • 1.995
