CRISPR vector construction with DNA assembly typically generates tens to hundreds of independent clones. Colony screening by PCR easily identifies correct clones and can distinguish between plasmids with and without inserts (Figure 2A) which is useful for troubleshooting. Typically, all of the clones contain an insert and a user may opt to skip the colony screening steps altogether. Diagnostic digests (Figure 2B) and Sanger sequencing are used for quality control. When errors do occur, they are usually observed in the overlapping DNA regions, i.e., the 5' end of the MtU6 promoter, the gRNA, or the 3' end of the scaffold sequence. If multiple gRNA oligos are pooled into a single reaction, the gRNAs are determined by Sanger sequencing. The incorporation of individual gRNAs is evenly distributed and most of the gRNAs can be isolated from a single transformation (Figure 2C). The high-fidelity DNA assembly mix is preferred over the standard DNA assembly mix as the high-fidelity version is less error-prone (Figure 2C).
Tomato hairy roots are usually observed one to two weeks after transformation (Figure 3A). Roots ≥2 cm in length are excised and transferred to selective medium and kanamycin-resistant roots can be identified one week later (Figure 3B). Most roots will grow to some degree on selective media and root growth in transformed roots can be variable; if possible, select the most vigorously growing roots for analysis. One to two kanamycin-resistant roots are typically recovered per cotyledon inoculated. With regular transfers, transformed roots can be maintained indefinitely on selective media.
DNA is extracted from kanamycin-resistant roots and PCR is used to amplify the target region(s). PCR products can be used in a variety of assays to determine mutation efficiency, such as cloning and sequencing (Figure 3C), restriction fragment length polymorphism analysis, polyacrylamide gel electrophoresis16 (Figure 3D), T7E1 endonuclease16, or high-throughput amplicon sequencing10. As reported in other CRISPR-plant systems6, depending on the gRNA/target sequence selected, a range of mutations and mutation efficiencies may be observed.
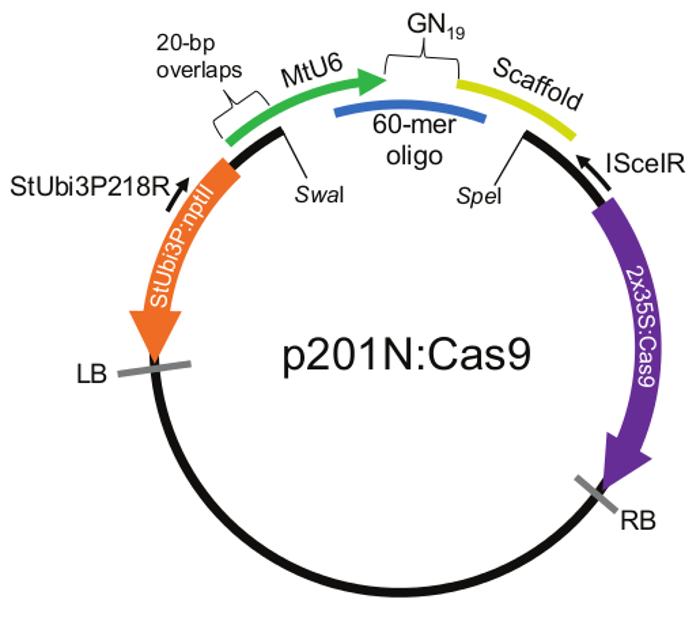
Figure 1. Principle of CRISPR Assembly. In the DNA assembly reaction, four DNAs are mixed together; p201N:Cas9 linearized with SwaI and SpeI (black circle), MtU6 promoter (green arrow), Scaffold (yellow bar) and the 60-mer gRNA oligo containing the GN19 target sequence (blue bar). The ends of each DNA molecule overlaps 20 bp with the adjacent piece. The DNA assembly reaction recombines these DNAs into a single, circular molecule. The StUbi3p218R and ISceIR primer binding sites are indicated with black arrows, the 2x35S:Cas9 cassette is purple, the StUbi3:nptII cassette is orange and left and right borders are indicated with grey bars. Note: Vector map is not to scale. Please click here to view a larger version of this figure.
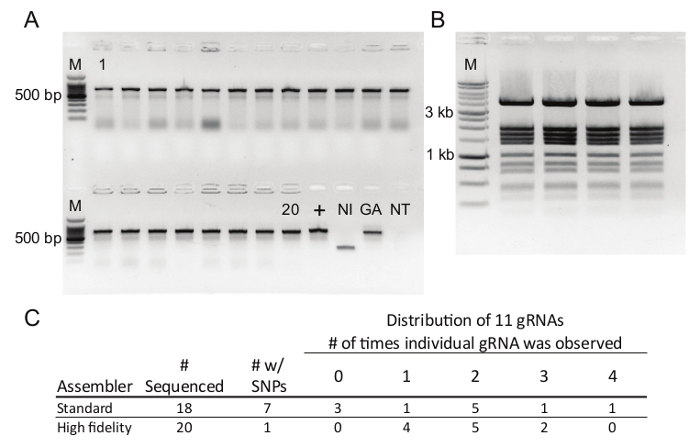
Figure 2. Colony Screening and Plasmid Quality Control. A) Colony screening of 20 clones from DNA assembly and transformation. M, 100-bp ladder; +, p201N:Cas9:GFPT2 positive control; NI, p201N:Cas9 no-insert control; GA, diluted DNA assembly reaction; NT, no-template control. B) Representative digest of four p201N:Cas9:gRNA vectors with the restriction enzymes EcoRV and StyI. M, 2-log ladder. C) Comparison of fidelity of the standard and high-fidelity DNA assembly mixes and distribution of recovered vectors when 11 gRNA oligos were pooled into a single reaction. Please click here to view a larger version of this figure.
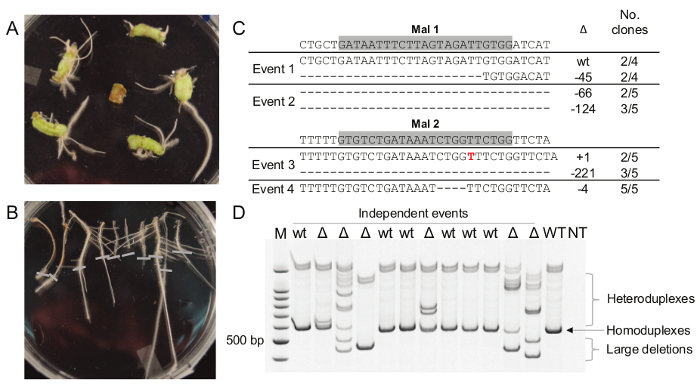
Figure 3. Hairy Root Cultures and Detection of DNA Modification. A) Hairy roots can be seen emerging from cotyledons 11 days after transformation. B) Roots are selected on ½ MS Kan50 media for approximately one week. Grey bars denote position of root tips at time of plating. C) Example of cloning and sequencing to determine DNA mutations with two different gene targets in four different hairy-root events. Grey box denotes GN20GG target sequence, Δ indicates type of mutation, and the number of clones with the indicated mutation. D) Example of polyacrylamide gel electrophoresis to determine DNA mutations. Large deletions and heteroduplex bands indicate the presence of DNA mutations at the target sequences. Six of 12 independent events were scored as mutants (Δ). WT, wild-type control; NT, no-template control. Please click here to view a larger version of this figure.
