Sequencing en genotypering op basis van studies, met inbegrip van Genome-Wide Association Studies (GWAS), kandidaat locus studies, en diep-sequencing studies, hebben veel genetische varianten die statistisch worden geassocieerd met een ziekte, trek, of fenotype geïdentificeerd. Anders dan vroege voorspellingen meeste van deze varianten (85-93%) zijn in niet-coderende gebieden en niet de aminozuursequentie van eiwitten 1,2 niet veranderen. De functie van deze niet-coderende varianten interpreteren en die de biologische mechanismen ze op de geassocieerde ziekte heeft eigenschap of fenotype bewezen uitdagende 3-6. genexpressie – We hebben een algemene strategie om de moleculaire mechanismen die varianten verwijzen naar een belangrijk tussenproduct fenotype te identificeren ontwikkeld. Deze leiding is speciaal ontworpen om modulatie van TF binding door genetische varianten te identificeren. Deze strategie combineert computationele benaderingen en moleculair biologische technieken om te voorspellenbiologische effecten van kandidaat-varianten in silico en controleer deze voorspellingen empirisch (figuur 1).
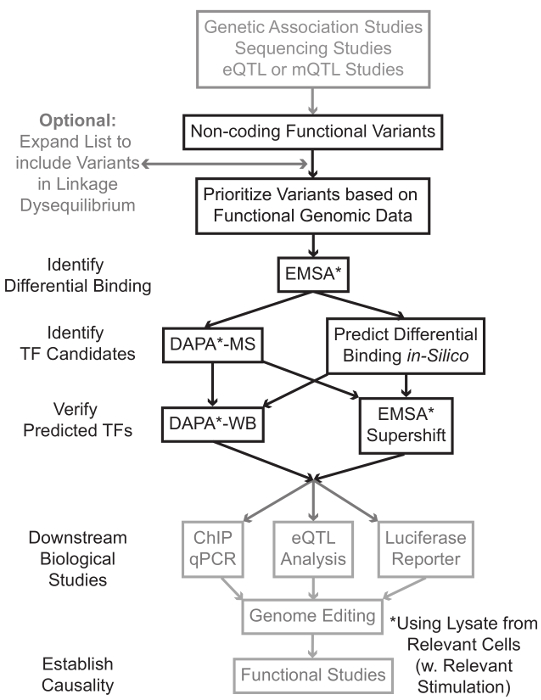
Figuur 1:.. Strategische aanpak voor de analyse van niet-coderende genetische varianten stappen die niet zijn opgenomen in de gedetailleerde protocol in verband met dit manuscript zijn grijs Klik hier om een grotere versie van deze figuur te bekijken.
In veel gevallen is het belangrijk om te beginnen uitbreiding van de lijst van varianten die allemaal in hoge linkage-onevenwicht (LD) met elkaar verbonden statistisch variant omvatten. LD is een maat voor niet-willekeurige associatie van allelen op twee verschillende chromosomale posities, die kan worden gemeten door de r 2 7 statistiek. r 2 is een maat voor de linKage onevenwichtigheid tussen twee varianten, met een r 2 = 1 aangeeft perfecte verbinding tussen twee varianten. Allelen hoge LD blijken co-segregeren op het chromosoom in voorouderlijke populaties. Huidige genotypering arrays niet alle bekende varianten in het menselijk genoom. In plaats daarvan gebruik maken van de LD in het menselijk genoom en omvatten een subset van de bekende varianten die als proxies voor andere varianten binnen een bepaalde regio van LD 8. Aldus kan een variant zonder biologische gevolg worden geassocieerd met een bepaalde ziekte omdat het in LD met de causale variant-de variant met een zinvolle biologisch effect. Procedureel, is het raadzaam om te zetten van de nieuwste versie van de 1000 genomen projecteren 9 variant call-bestanden (VCF) in binaire bestanden compatibel zijn met Plink 10,11, een open-source tool voor het gehele genoom vereniging analyse. Vervolgens worden alle andere genetische varianten met LD r 2> 0,8 bij elke ingang genetische variant kunnen worden geïdentificeerd als kandidaten. Het is belangrijk om de overeenkomstige referentiegroep voor dit stap- bijvoorbeeld als een variant geïdentificeerd bij personen van Europese afkomst, de data van onderwerpen van soortgelijke afkomst moeten worden gebruikt voor LD expansie.
LD expansie resulteert vaak in tientallen kandidaat varianten, en het is waarschijnlijk dat slechts een klein deel daarvan bijdragen aan ziektemechanisme. Vaak is onhaalbaar experimenteel onderzoeken elk van deze varianten afzonderlijk. Daarom is het nuttig om de duizenden publiekelijk beschikbare functionele genomische datasets te benutten als een filter om voorrang te geven aan de varianten. Zo heeft de ENCODE consortium 12 duizenden ChIP-seq experimenten waarin de binding van TF en co-factoren en histon markeringen uitgevoerd in diverse contexten, samen met chromatine bereikbaarheidsgegevens van technologieën zoals DNase-seq 13, ATAC -seq 14 en FAIRE-seq 15. databases en webservers zoals de UCSC Genome Browser 16, Roadmap Epigenomics 17, Blueprint Epigenome 18, Cistrome 19 en Remap 20 bieden gratis toegang tot de gegevens die door deze en andere experimentele technieken in een breed scala van celtypen en voorwaarden. Wanneer er te veel varianten experimentele onderzoeken, kunnen deze gegevens worden gebruikt om prioriteit die zich binnen waarschijnlijk regulerende gebieden desbetreffende cel- en weefseltypen. Verder wanneer een variant binnen een ChIP-seq piek voor een specifiek eiwit, deze gegevens kunnen potentiële leads geven over de specifieke TF (s) of co-factoren waarvan de binding zou kunnen beïnvloeden.
Vervolgens worden de resulterende prioriteit varianten experimenteel gescreend om voorspelde genotype-afhankelijke eiwit binding te valideren met behulp van EMSA 21,22. EMSA meet de verandering in de migratie van de oligo op een niet-reducerende TBE gel. Fluorescent gemerkte oligo wordt geïncubeerd met denucleaire lysaat en binding van nucleaire factoren de beweging van de oligo vertragen op de gel. Op deze wijze oligo dat meer kernfactoren is gebonden zal een sterker fluorescentiesignaal op scantoepassingen. Met name EMSA geen voorspellingen over de specifieke eiwitten waarvan de binding zal worden beïnvloed vereisen.
Zodra varianten zijn geïdentificeerd die zich binnen het voorspelde regelende gebieden en kunnen verschillend binding kernfactoren zijn computationele werkwijzen toegepast om de specifieke TF (n) waarvoor bindende ze invloed kunnen voorspellen. Wij geven de voorkeur aan CIS-BP 23,24, RegulomeDB 25, UniProbe 26 en JASPAR 27 gebruiken. Zodra de kandidaat-TF's worden geïdentificeerd, kunnen deze voorspellingen specifiek worden getest met behulp van antilichamen tegen deze TF's (EMSA-supershifts en DAPA-Westerns). Een EMSA-supershift bij de bereiding een TF-antilichamen voor de nucleaire lysaat en oligo. Een positief resultaat in een EMSA-supershift is represented een verdere verschuiving in de EMSA band, of een verlies van de band (besproken in referentie 28). In de complementaire DAPA, een 5'-gebiotinyleerde oligo duplex die de variant en 20 basenparen flankerende nucleotiden zijn geïncubeerd met nucleaire lysaat uit relevante type (s) cel nucleaire factoren die specifiek binden de oligo vangen. De duplex-oligo nucleaire factor complex geïmmobiliseerd op streptavidine microkralen in een magnetische kolom. De gebonden nucleaire factoren worden rechtstreeks verzameld via elutie 29,48. Bindende voorspellingen kunnen dan worden beoordeeld door een Western blot met gebruik van antilichamen specifiek voor het eiwit. In gevallen waarin er geen duidelijke voorspellingen, of te veel voorspellingen, de eluties van variant pull-downs van de DAPA experimenten kan een proteomics kern worden gestuurd om de kandidaat-TF's te identificeren met behulp van massaspectrometrie, die vervolgens kunnen worden gevalideerd met behulp van deze eerder beschreven werkwijzen.
In de rest van de article, is de gedetailleerde protocol voor het EMSA en DAPA analyse van genetische varianten voorzien.
