Sequenzierung und Genotypisierung basierte Studien, einschließlich der genomweiten Assoziationsstudien (GWAS), Kandidaten-Locus Studien und tiefSequenzierungsUntersuchungen haben viele genetische Varianten identifiziert, die statistisch im Zusammenhang mit einer Krankheit, Zug oder Phänotyp. Im Gegensatz zu dem frühen Vorhersagen, die meisten dieser Varianten (85-93%) werden in nicht-codierenden Regionen und die Aminosäuresequenz von Proteinen nicht 1,2 ändern. Interpretieren der Funktion dieser nicht-kodierenden Varianten und Bestimmung der biologischen Mechanismen , um sie zu dem zugeordneten Krankheit verbindet, trait oder Phänotyp hat Herausforderung erwiesen 3-6. Wir haben eine allgemeine Strategie entwickelt, um die molekularen Mechanismen zu identifizieren, die Varianten zu einem wichtigen Zwischen Phänotyp verknüpfen – Genexpression. Diese Pipeline wird speziell zu identifizieren, die Modulation der TF-Bindung durch genetische Varianten entwickelt. Diese Strategie kombiniert Rechenansätze und Techniken der Molekularbiologie Ziel vorherzusagenbiologische Wirkungen von Kandidatenvarianten in silico, und überprüfen diese Prognosen empirisch (Abbildung 1).
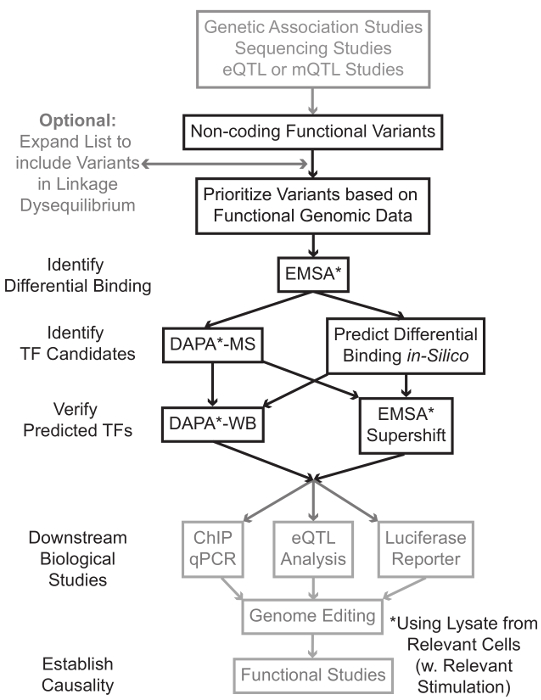
Abbildung 1:.. Strategischer Ansatz für die Analyse von nicht-kodierenden genetischen Varianten Schritte, die in der detaillierten Protokoll sind nicht mit diesem Manuskript assoziiert enthalten sind grau schattiert Bitte hier klicken , um eine größere Version dieser Figur zu sehen.
In vielen Fällen ist es wichtig, durch eine Erweiterung der Liste der Varianten zu beginnen alle, die in hohen Gestänge-Ungleichgewichts (LD) mit jeder statistisch assoziiert Variante enthalten. LD ist ein Maß der nicht-zufällige Assoziation von Allelen an zwei verschiedenen chromosomalen Stellen, die durch die r 2 -Statistik 7 gemessen werden kann. r 2 ist ein Maß für die linKage Ungleichgewichts zwischen zwei Varianten, mit einem r 2 = 1 bezeichnet perfekte Verbindung zwischen zwei Varianten. Allelen in hohen LD gefunden auf dem Chromosom über Vorfahren Populationen zusammen entmischen. Aktuelle Genotypisierung Arrays enthalten nicht alle bekannten Varianten im menschlichen Genom. Stattdessen verwerten sie die LD innerhalb des menschlichen Genoms und sind eine Teilmenge der bekannten Varianten , die 8 für andere Varianten innerhalb einer bestimmten Region des LD als Stellvertreter fungieren. Somit kann mit einer bestimmten Krankheit assoziiert sein eine Variante ohne biologische Folge kann, da es in LD mit dem kausalen varianten der Variante mit einer sinnvollen biologischen Wirkung. Prozedural, wird empfohlen , die neueste Version der 1000 Genome Projekt 9 Variante Call – Dateien (VCF) in binäre Dateien zu konvertieren kompatibel mit PLINK 10,11, einem Open-Source – Tool für Genom – Assoziations – Analyse. Anschließend werden alle anderen genetischen Varianten mit LD r 2> 0,8 mit jedem Eingang genetischen variant als Kandidaten identifiziert werden. Es ist wichtig , die entsprechende Referenzpopulation für diese Schritt- zB zu verwenden, wenn eine Variante , bei Themen von europäischer Abstammung identifiziert wurde, Daten von Patienten mit ähnlicher Herkunft sollte für LD Expansion verwendet werden.
LD Expansion resultiert oft in Dutzenden von Kandidatenvarianten, und es ist wahrscheinlich, dass nur ein kleiner Bruchteil von diesen zu Krankheitsmechanismus beizutragen. Oft ist es nicht durchführbar, experimentell jede dieser Varianten einzeln untersuchen. Es ist daher sinnvoll, die Tausende von öffentlich verfügbaren funktionellen genomischen Datensätze als Filter zu nutzen, um die Varianten zu priorisieren. Zum Beispiel 12 das ENCODE – Konsortium hat Tausende von ChIP-seq Experimente beschreiben die Bindung von Transkriptionsfaktoren und Co-Faktoren, und Histonmarkierungen in einem breiten Spektrum von Kontexten, zusammen mit Chromatin Zugänglichkeit von Daten von Technologien wie DNase-seq 13, ATAC ausgeführt -SEQ 14 und FAIRE-seq 15. DatabAses und Web – Servern wie dem UCSC Genome Browser 16, Roadmap Epigenomics 17, Epigenome Blueprint 18, Cistrome 19 und ReMap 20 bieten freien Zugang zu Daten , die durch diese und andere experimentelle Techniken in einer Vielzahl von Zelltypen und Bedingungen hergestellt. Wenn es zu viele Varianten sind experimentell zu untersuchen, können diese Daten verwendet werden, die innerhalb wahrscheinlich regulatorischen Regionen in entsprechenden Zell- und Gewebetypen angeordnet zu priorisieren. Ferner wird in Fällen, in denen eine Variante für ein spezifisches Protein in einer ChIP-seq peak ist, können diese Daten Potential führt als der spezifischen TF (s) oder Cofaktoren schaffen, deren Bindung könnte beeinflussen.
Als nächstes werden die resultierenden priorisiert Varianten gescreent experimentell vorhergesagt Genotyp-abhängige Protein zu validieren Bindungs 21,22 mittels EMSA. EMSA misst die Veränderung in der Migration des oligo auf einem nicht-reduzierenden TBE-Gel. Fluoreszierend markiertes Oligo mit der inkubiertenKern Lysat, und die Bindung von Kernfaktoren wird verzögern die Bewegung des Oligo auf dem Gel. Auf diese Weise Oligo, die mehr Kernfaktoren gebunden hat, als ein stärkeres Fluoreszenzsignal beim Abtasten präsentieren wird. Bemerkenswert ist, erfordert keine EMSA Prognosen über die spezifischen Proteine, deren Bindung betroffen.
Sobald Varianten identifiziert werden, die innerhalb der erwarteten regulatorischen Regionen befinden und in der Lage sind unterschiedlich Bindung Kernfaktoren sind Berechnungsverfahren die spezifische TF (s), dessen vorherzusagen verwendet Bindung sie beeinflussen könnten. Wir bevorzugen CIS-BP 23,24, RegulomeDB 25, UNIProbe 26 zu verwenden, und JASPAR 27. Sobald Kandidaten TFs identifiziert werden, können diese Prognosen ausdrücklich Antikörper gegen diese TFs (EMSA-supershifts und DAPA-Westerns) getestet werden. Eine EMSA-Supershift beinhaltet die Zugabe eines TF-spezifischen Antikörpers an das Kern Lysat und Oligo. Ein positives Ergebnis in einem EMSA-Supershift ist represented als eine weitere Verschiebung der EMSA Band oder einem Verlust des Bandes (in Bezug 28 überprüft). In der komplementären DAPA, eine 5'-biotinylierten Oligokomplexes die Variante und das 20 Basenpaar-flankierenden Nukleotide enthalten, werden mit nuklearen Lysat von relevanten Zelltyp (en) inkubiert alle Kernfaktoren speziell zur Erfassung der Oligos binden. Die Oligokomplexes-nuclear factor-Komplex wird durch Streptavidin-Mikrokügelchen in einer magnetischen Säule immobilisiert. Die gebundenen Kernfaktoren werden direkt durch Elution 29,48 gesammelt. Bindungsvorhersagen können dann durch Western-Blot untersucht werden, um Antikörper, spezifisch für das Protein verwendet wird. In Fällen, in denen es keine offensichtlichen Prognosen oder zu viele Vorhersagen, die Elutionen von Variante Pull-downs der DAPA Experimente können zu einem Proteomik Kern gesendet werden, um Kandidaten TFs mit Massenspektrometrie identifizieren, die anschließend validiert werden können mit diesen zuvor beschriebenen Methoden.
In dem Rest des article wird das detaillierte Protokoll für EMSA und DAPA Analyse genetischer Varianten vorgesehen.
