Sequencing e studi di genotipizzazione basato, compresi gli studi Genome-Wide Association (GWAS), studi locus candidato, e profondo-sequenziamento studi, hanno identificato molte varianti genetiche che sono statisticamente associati con una malattia, caratteristica, o fenotipo. Contrariamente alle previsioni iniziali, la maggior parte di queste varianti (85-93%) si trovano in regioni non codificanti e non cambiano la sequenza di amminoacidi delle proteine 1,2. Interpretando la funzione di queste varianti non codificanti e determinare i meccanismi biologici che li collegano alla malattia associata, tratto, o fenotipo è dimostrato impegnativo 3-6. Abbiamo sviluppato una strategia generale di identificare i meccanismi molecolari che collegano le varianti di un importante fenotipo intermedio – l'espressione genica. Questo gasdotto è specificamente progettato per identificare la modulazione di legare da varianti genetiche TF. Questa strategia combina approcci computazionali e tecniche di biologia molecolare finalizzate a prevedereeffetti biologici delle varianti candidati in silico e verificare empiricamente queste previsioni (Figura 1).
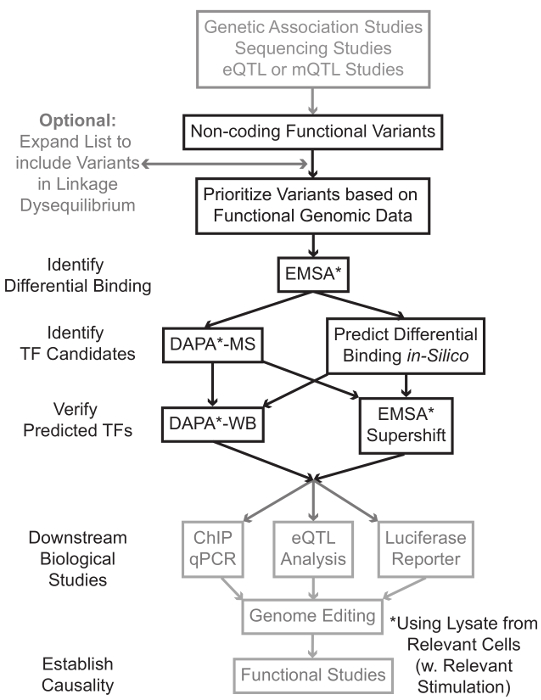
Figura 1:.. Approccio strategico per l'analisi dei passaggi non codificanti varianti genetiche che non sono inclusi nel protocollo dettagliato associato a questo manoscritto sono di colore grigio Clicca qui per vedere una versione più grande di questa figura.
In molti casi, è importante iniziare ampliare l'elenco delle varianti di includere tutti quelli in alta linkage disequilibrium (LD) con ciascuna variante statisticamente associato. LD è una misura di associazione non casuale di alleli in due differenti posizioni cromosomiche, che può essere misurata dal r 2 statistica 7. r 2 è una misura del linKage squilibrio fra due varianti, con una R 2 = 1 denota perfetto collegamento tra due varianti. Alleli in alta LD si trovano a co-separare sul cromosoma attraverso popolazioni ancestrali. array di genotipizzazione attuali non includono tutte le varianti conosciute nel genoma umano. Invece, sfruttano il LD all'interno del genoma umano e comprendono un sottoinsieme delle varianti note che agiscono come proxy per altre varianti di una particolare regione del LD 8. Così, una variante senza alcuna conseguenza biologica può essere associato ad una particolare malattia perché è in LD con causale variant-variante con un effetto biologico significativo. Procedurale, si consiglia di convertire l'ultima versione dei 1.000 genomi Project 9 file di chiamata variante (VCF) in file binari compatibili con PLINK 10,11, uno strumento open-source per l'intera analisi di associazione genoma. In seguito, tutte le altre varianti genetiche con LD r 2> 0.8 con ogni ingresso va geneticariant può essere identificato come candidati. E 'importante usare la popolazione di riferimento appropriato per questo passo- esempio, se una variante è stato identificato nei soggetti di origine europea, i dati provenienti da soggetti di origini simili dovrebbero essere utilizzati per l'espansione LD.
espansione LD si traduce spesso in decine di varianti candidati, ed è probabile che solo una piccola frazione di queste contribuiscono alla malattia meccanismo. Spesso, è praticamente impossibile esaminare sperimentalmente ciascuna di queste varianti singolarmente. E 'quindi utile per sfruttare le migliaia di pubblicamente disponibili i set di dati di genomica funzionale come filtro per dare priorità alle varianti. Ad esempio, il consorzio ENCODE 12 ha eseguito migliaia di esperimenti di ChIP-Seq che descrivono il legame del TF e co-fattori, e segni istoni in una vasta gamma di contesti, insieme ai dati della cromatina di accessibilità da tecnologie come DNasi-ss 13, ATAC -seq 14, e FAIRE-ss 15. databAsi e server web, come il browser UCSC Genome 16, tabella di marcia Epigenomics 17, Blueprint Epigenome 18, Cistrome 19, e Remap 20 fornire libero accesso ai dati prodotti da questi e altre tecniche sperimentali in una vasta gamma di tipi e condizioni di cellule. Quando ci sono troppe varianti per esaminare sperimentalmente, questi dati possono essere utilizzati per dare la priorità quelli situati all'interno probabili regioni regolatorie in materia tipi di cellule e tessuti. Inoltre, nei casi in cui una variante è all'interno di un picco ChIP-seq per una proteina specifica, questi dati possono fornire cavi potenziali per il TF specifico (s) o cofattori cui legame potrebbe essere che interessano.
Successivamente, le varianti risultanti priorità sono proiettati sperimentale per validare proteina genotipo-dipendente predetto legame con EMSA 21,22. EMSA misura la variazione della migrazione del oligo su un gel TBE non riducente. oligo fluorescente è incubato con illisato nucleare e legame di fattori nucleari ritardare il movimento del oligo sul gel. In questo modo, oligo che ha legato più fattori nucleari presenterà come un segnale fluorescente forte alla scansione. In particolare, l'EMSA non richiede previsioni circa le proteine specifiche il cui legame saranno interessati.
Una volta che le varianti sono identificati che si trovano all'interno di regioni regolatorie previsti e sono in grado di fattori nucleari differenziale vincolanti, metodi computazionali sono impiegati per predire il TF specifico (s), il cui legame che potrebbero influenzare. Noi preferiamo usare CIS-BP 23,24, RegulomeDB 25, UNIProbe 26, e JASPAR 27. Una volta che il candidato TF sono identificate, queste previsioni possono essere specificamente testati utilizzando anticorpi contro questi TF (EMSA-supershifts e DAPA-western). Un EMSA-supershift comporta l'aggiunta di un anticorpo specifico TF al lisato nucleare e oligo. Un risultato positivo in un EMSA-supershift è represented come un ulteriore spostamento nella banda EMSA, o una perdita di banda (valutata in riferimento 28). Nel DAPA complementare, un duplex oligo 5'-biotinilato contenente la variante e il 20 paia di basi di accompagnamento nucleotidi sono incubati con lisato nucleare dal tipo di cellula in questione (s) per catturare eventuali fattori nucleari legame specifico gli oligonucleotidi. Il complesso fattore duplex-nucleare oligo è immobilizzato dalla streptavidina microsfere in una colonna magnetica. I fattori nucleari legati vengono raccolti direttamente tramite eluizione 29,48. previsioni vincolanti possono poi essere valutati da un Western blot utilizzando anticorpi specifici per la proteina. Nei casi in cui non ci sono previsioni evidenti, o anche molte previsioni, i eluizioni dalla variante di pull-down degli esperimenti DAPA possono essere inviati a un nucleo di proteomica per identificare TF candidati mediante spettrometria di massa, che possono in seguito essere convalidati utilizzando questi precedentemente descritte metodi.
Nel resto della articlE, viene fornito il protocollo dettagliato per EMSA e DAPA analisi delle varianti genetiche.
