Sekvensering och genotypning baserade studier, inklusive Genomvid associationsstudier (GWAS), kandidat locus studier och djupsekvense studier har identifierat många genetiska varianter som statistiskt är associerade med en sjukdom, drag eller fenotyp. I motsats till tidigare prognoser, de flesta av dessa varianter (85-93%) är belägna i icke-kodande regioner och ändrar inte aminosyrasekvensen av proteiner 1,2. Tolka funktion av dessa icke-kodande varianter och bestämma de biologiska mekanismerna som förbinder dem till associerad sjukdom, har drag, eller fenotyp visat utmanande 3-6. Vi har utvecklat en övergripande strategi för att identifiera de molekylära mekanismer som länkar varianter till en viktig mellan fenotyp – genuttryck. Denna pipeline är särskilt utformade för att identifiera moduleringen av TF bindning av genetiska varianter. Denna strategi kombinerar beräkningsmetoder och molekylärbiologiska tekniker som syftar till att förutsägabiologiska effekter av kandidat varianter i silico och verifiera dessa förutsägelser empiriskt (Figur 1).
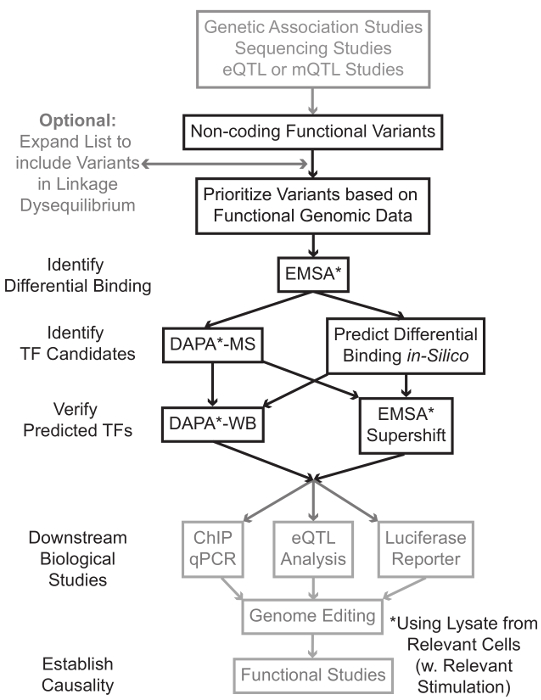
Figur 1:.. Strategi för analys av icke-kodande genetiska varianter Steg som inte ingår i den detaljerade protokoll i samband med detta manuskript är skuggade i grått Klicka här för att se en större version av denna siffra.
I många fall är det viktigt att börja med att utöka listan över varianter att omfatta alla de i hög linkage-ojämvikt (LD) med varje statistiskt samband variant. LD är ett mått på icke-slumpmässigt association av alleler vid två olika kromosomala positioner, som kan mätas av r 2 statistik 7. r 2 är ett mått på linKage obalans mellan två varianter, med en r 2 = 1 betecknar perfekt koppling mellan två varianter. Alleler i hög LD befinns samtidigt segregera på kromosomen över släkt- befolkningar. Nuvarande genotypning arrayer inkluderar inte alla kända varianter i det mänskliga genomet. Istället utnyttjar LD inom det mänskliga genomet och inkludera en delmängd av de kända varianter som fungerar som ombud för andra varianter inom en viss region i LD 8. Således kan en variant utan någon biologisk konsekvens vara associerad med en särskild sjukdom eftersom det är i LD med orsaks variant-varianten med en meningsfull biologisk effekt. Procedurmässigt är det rekommenderat att konvertera den senaste versionen av 1000 genomen projektet 9 variant samtals filer (VCF) till binära filer som är kompatibla med Plink 10,11, ett open-source verktyg för hela genomet associationsanalys. Därefter alla andra genetiska varianter med LD r 2> 0,8 med varje ingång genetisk variant kan identifieras som kandidater. Det är viktigt att använda den tillämpliga referenspopulationen för denna steg t ex om en variant identifierades i frågor av europeisk härkomst, data från patienter med liknande härkomst ska användas för LD expansion.
LD expansionen resulterar ofta i dussintals kandidat varianter, och det är troligt att endast en liten del av dessa bidrar till sjukdomsmekanismen. Ofta är det omöjligt att experimentellt undersöka var och en av dessa varianter individuellt. Det är därför lämpligt att utnyttja de tusentals offentligt tillgängliga funktionsgenomiska datamängder som ett filter för att prioritera varianter. Till exempel har ENCODE konsortiet 12 utfört tusentals Chip-punkter experiment som beskriver bindningen av TF och co-faktorer, och histon märken i en rad olika sammanhang, tillsammans med kromatin tillgänglighetsdata från teknik såsom DNas-punkter 13, ATAC -seq 14, och FAIRE-seq 15. DatabALL och webbservrar som UCSC Genome Browser 16, färdplan Epigenomics 17, Blueprint Epigenome 18, Cistrome 19, och mappa 20 ger fri tillgång till data som produceras av dessa och andra experimentella metoder inom ett brett spektrum av celltyper och villkor. När det finns för många varianter att undersöka experimentellt, kan dessa data användas för att prioritera de som ligger inom troliga reglerande regioner inom relevanta cell- och vävnadstyper. Vidare, i de fall där en variant inom en chip-punkter topp för ett specifikt protein, kan dessa uppgifter ger potentiella kunder som den specifika TF (s) eller co-faktorer vars bindning kan påverka.
Därefter de resulterande prioriterade varianter screenas experimentellt att validera förutspått genotyp beroende proteinbindning med hjälp av EMSA 21,22. EMSA mäter förändringar i migreringen av oligo på en icke-reducerande TBE-gel. Fluorescensmärkt oligo inkuberas mednukleära lysat, och bindning av nukleära faktorer kommer att fördröja rörelsen av oligo på gelén. På detta sätt, oligo som har bunden fler nukleära faktorer kommer att presentera som en starkare fluorescerande signal vid scanning. Noterbart är inte EMSA inte kräva förutsägelser om de specifika proteiner vars bindning kommer att påverkas.
När varianter identifieras som är belägna inom förutsedda regulatoriska regioner och är i stånd att differentiellt bindande kärn faktorer, är beräkningsmetoder som används för att förutsäga specifika TF (s) vars bindande de kan påverka. Vi föredrar att använda CIS-BP 23,24, RegulomeDB 25 UniProbe 26, och Jaspar 27. När kandidat TF har identifierats, kan dessa förutsägelser specifikt testas med hjälp av antikroppar mot dessa TF (EMSA-supershifts och DAPA-Westerns). En EMSA-supershift innebär tillsättning av en TF-specifik antikropp mot den nukleära lysat och oligo. Ett positivt resultat i en EMSA-supershift är represented som en ytterligare förskjutning i EMSA-bandet, eller en förlust av bandet (översikt i referens 28). I den komplementära DAPA är en 5'-biotinylerad oligo duplex innehållande varianten och 20 baspar som flankerar nukleotider inkuberas med kärn lysat från relevant celltyp (s) för att fånga några nukleära faktorer som specifikt binder oligos. Oligo duplex-nukleär faktor komplexet immobiliseras genom streptavidin mikropärlor i en magnetisk kolonn. De bundna nukleära faktorer samlas direkt genom eluering 29,48. Bindande prognoser kan sedan bedömas av en Western blöt med användning av antikroppar som är specifika för proteinet. I de fall där det inte finns några uppenbara förutsägelser, eller alltför många förutsägelser, de elueringar från variantrullgardinsmenyerna av DAPA experiment kan skickas till en proteomik kärna för att identifiera kandidat TF använder masspektrometri, som därefter kan valideras med hjälp av dessa tidigare beskrivna metoder.
I återstoden av article är den detaljerade protokoll för EMSA och DAPA analys av genetiska varianter tillhandahålls.
