Unvoreingenommene Tiefe Sequenzierung von RNA-Viren aus klinischen Proben
Summary
This protocol describes a rapid and broadly applicable method for unbiased RNA-sequencing of viral samples from human clinical isolates.
Abstract
Hier beschreiben wir eine Next-Generation – RNA – Sequenzierung Protokoll , das de novo Assemblierung und intra-Host – Variante Anrufe von viralen Genomen von klinischen und biologischen Quellen gesammelt werden können. Die Methode ist unvoreingenommen und universell; es verwendet zufällige Primer für die cDNA-Synthese und erfordert keine vorherige Kenntnis der viralen Sequenz Gehalt. Vor dem Aufbau der Bibliothek, selektive RNase H-basierte Verdauung wird verwendet, um unerwünschte RNA zu führen – einschließlich Poly (rA) Träger und ribosomale RNA – von der viralen RNA-Probe. Selective Depletion verbessert sowohl die Datenqualität und die Anzahl der eindeutigen liest in der viralen RNA-Sequenzierung Bibliotheken. Darüber hinaus ist ein Transposase-basierten 'tagmentation' Schritt wird in dem Protokoll verwendet wird, wie es allgemeine Bibliothek Bauzeit reduziert. Das Protokoll hat eine rasante tiefe Sequenzierung von mehr als 600 Lassa und Ebola-Virusproben-Sammlungen einschließlich aktiviert sowohl von Blut und Gewebe isoliert-und ist im Großen und Ganzen auf andere mikrobielle Genomstudien.
Introduction
Next Generation Sequenzierung von Viren aus klinischen Quellen kann die Übertragung und die Epidemiologie von Infektionen informieren, sowie dazu beitragen, Unterstützung neuer diagnostischer, Impfstoff und therapeutische Entwicklung. cDNA – Synthese unter Verwendung von Zufallsprimer hat den Nachweis und die Montage von Genomen von divergent, Coinfizierung oder auch neuartige Viren 1,2 erlaubt. Wie bei anderen unvoreingenommene Methoden, besetzen unerwünschte Verunreinigungen viele Sequenzierung liest und negativ Sequenzierungsergebnisse auswirken. Host-und Poly (rA) Carrier-RNA sind Verunreinigungen, die in vielen bestehenden viralen Musterkollektionen.
Das Protokoll beschreibt eine effiziente und kostengünstige Möglichkeit der tiefen Sequenzierung Genomen RNA Virus basiert auf unvoreingenommene total RNA-seq. Das Verfahren nutzt eine RNase H selektive Depletion Schritt 3 unerwünschte Wirt ribosomalen und Carrier – RNA zu entfernen. Selektive Depletion reichert für virale Gehalt (Abbildung 1) und verbessert die Gesamtqualität der Sequenzierungsdaten(Abbildung 2) aus klinischen Proben. Außerdem wird tagmentation dem Protokoll angewendet, wie es deutlich Bibliothek Bauzeit reduziert. Diese Methoden wurden verwendet , um schnell große Datenmengen von Ebola und Lassa – Virus – Genome 2,4,5 erzeugen und kann verwendet werden , um ein breites Spektrum von RNA – Viren zu untersuchen. Schließlich wird der Ansatz nicht auf menschlichen Proben beschränkt; Die Nützlichkeit der selektive Depletion wurde auf Gewebeproben von Lassa-infizierte Nager und nicht-menschlichen Primaten – Krankheitsmodellen 5,6 gesammelt gezeigt.
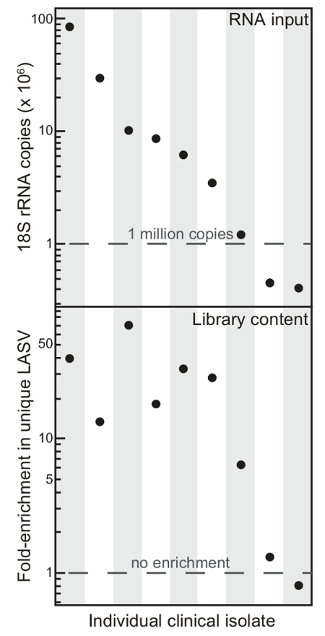
Abbildung 1. Die Gesamt – RNA Inhalt Reflektiert Anreicherung von Lassa – Virus von Inhalt mit Selective Depletion. Beginnend Gesamtinhalt (RNA – Eingang) und Anreicherung von einzigartigen Lassa – Virus (LASV) liest (Library Inhalt) auf rRNA Depletion von neun verschiedenen klinischen Isolaten. Diese Zahl wurde von 6 geändert <em>. Bitte hier klicken , um eine größere Version dieser Figur zu sehen.
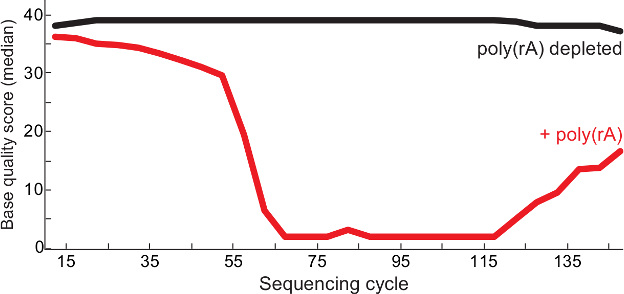
Abbildung 2. Höhere Qualität Sequenzierung Nach Carrier – RNA Depletion. Median Basisqualitäten pro Sequenzierzyklus von Poly (rA) -contaminated Bibliotheken Lassa – Virus (rot) und Kontrolle (kein Träger in der Bibliothek beobachtet, schwarz) von QC – Bericht 13. Beide lesen 1 und 2 gelesen gepaarter Ende liest in der Bibliothek BAM-Datei zusammengefügt und die Qualitätswerte werden an jeder Basis gezeigt. Diese Zahl hat sich von 6 geändert. Bitte klicken Sie hier , um eine größere Version dieser Figur zu sehen.
Die virale RNA-seq Protokoll Details Bau von Bibliotheken direkt aus extrahiertenRNA gesammelt aus klinischen und biologischen Proben. Um sicherzustellen, dass die persönliche Sicherheit, alle viralen Serum, Plasma und Gewebeproben sollten in geeigneten Puffern vor der RNA-Extraktion inaktiviert werden. In einigen Inaktivierung und Extraktion-Kits, Träger Poly (rA) RNA ist im Preis inbegriffen; Dies wird während der anfänglichen RNase H selektive Depletion Schritt entfernt werden. Basierend auf vollständige Genesung, ist die erwartete Konzentration von Carrier-RNA 100 ng / ul. In dem Protokoll, 110 ng / & mgr; l Oligo-dT-RNA (1.1x Ladungsträgerkonzentration), zur Abreicherung eingesetzt. Wenn Poly (rA) Träger nicht in der Probe vorhanden ist, dann Oligo (dT) nicht hinzugefügt, um Erschöpfung vor werden.
Das folgende Protokoll wird für 24 Reaktionen in PCR-Plattenformat ausgelegt (bis zu 250 & mgr; l Volumen). Eine frühere Version dieses Protokolls wurde in Matranga, et al. 6.
Protocol
Representative Results
Discussion
Das skizzierte Ansatz ermöglicht robuste, universelle, schnelle Sequenzierung und wurde zur Sequenzierung von Ebola – Virus während der 2014 Ausbruch 2,4 verwendet. Durch die Kopplung selektive Depletion und cDNA-Synthese mit tagmentation Bibliothekskonstruktion wurde die Gesamtprozesszeit um ~ 2 Tage reduziert von früheren Methoden Adaptorligation. In jüngerer Zeit wurde dieses Protokoll , das von internationalen Mitarbeiter und andere mit großem Erfolg 15,16 und wird eingesetzt , um Labors in Westafrika zu unterstützen lokale Genomik-basierte Studien und Diagnostik 17 eingesetzt.
Das hier beschriebene Protokoll verwendet Zufallsprimer cDNA-RNA-seq-Bibliotheken für die virale vorzubereiten. Im Gegensatz zu früheren viralen RNA-seq Ansätze, es erfordert keine a priori Kenntnis von Sequenzdaten oder aufwendige und zeitraubende Primerdesign für einen bestimmten Virus oder Klade. Das Verfahren kann auf jede viral RNA Probe angewendet werden. Beispielsweise wurde es verwendet, viralen Gehalt von sowohl Ebola zu erzeugenund Lassa Proben 6. Das Protokoll kann auch Projekte für Host transkriptomischen, Metagenom und Erreger Entdeckung Sequenzierung 1 verwendet werden.
Ein kritischer Schritt des Protokolls wird RNase H Verdau gezielte, ein Hochdurchsatz, kostengünstiges Verfahren zum Entfernen von unerwünschten Träger- und Host-RNA aus viralen Proben. Die selektive Depletion Schritt des Protokolls verwendet viele Komponenten und erfordert Geschicklichkeit und Genauigkeit. Extra-Zeit und Sorgfalt sollte bei der Ersteinrichtung genommen werden.
Da die meisten klinischen Serum- und Plasmaproben oft sehr wenig Nukleinsäurematerial aufweisen, sind Verunreinigungen und Probenverlust gemeinsam. Um diese Probleme zu vermeiden, sollten Sie besonders vorsichtig sein, wenn dieses Protokoll verwenden. Zunächst wird RNA sehr anfällig für Degradation; Daher sollten alle Bereiche sauber und frei von Nukleasen sein. Zweitens, um Proben zu identifizieren geeignet für den Einsatz in diesem Protokoll qRT-PCR – Assays für sowohl Wirts – RNA und Virus sollte zur Quantifizierung 5,6 verwendet werden </sup>. Wenn der Eingang Vergleich beträgt mit Sequenzierung ergibt sich aus dem Protokoll, Sequenzierung Erfolg (dh die Erzeugung von genügend Daten für die vollständige virale Montage) korreliert mit Proben , die mindestens 100 pg Gesamt – RNA und 1.000 Kopien des Virus enthalten. Drittens Einwirkung von Umweltquellen von Nukleinsäuren sollten vermieden werden. Das Protokoll hier skizzierte in einem Biosicherheitsschrank für Sicherheitsvorkehrungen durchgeführt und zur Begrenzung der Umweltschadstoffe. Darüber hinaus haben unsere Gruppe und anderen bemerkt , dass kommerzielle Enzyme bakterielle Nukleinsäuren in niedrigen Eingangsproben von verunreinigenden 6,18 eine andere Quelle sein kann. Die Verwendung eines sauberen Arbeitsplatz (zB PCR – Haube, biologische Sicherheit Kabinett) und Negativkontrollen (zB Wasser oder Puffer) wird dazu beitragen , lindern und Verschmutzung zu verfolgen sind. Bei Proben mit <100 pg Gesamt-RNA, nur Poly (rA) Carrier-RNA, nicht rRNA, sollte eine hohe Qualität Sequenzierergebnisse erschöpft sein, um sicherzustellen, während Materialverlust zu begrenzen. Für sehrniedrige Eingangsproben cDNA-Amplifikationsverfahren können 19 besser geeignet sein, obwohl Poly (rA) Träger sollte vor der cDNA – Synthese entfernt werden.
Die Abreicherung von Wirts rRNA reichert für virale Gehalt in Sequenzierungsbibliotheken und ist anwendbar auf verschiedene Probensammlungen einschließlich Serum oder Plasma, und mehrere Arten von Gewebe von Nagern und nicht-menschlichen Primaten 5,6. In nicht-menschlichen Organismen, liest Ausrichten zu 28S rRNA nach Erschöpfung blieb, was darauf hindeutet 28S rRNA ist weniger konserviert zwischen Menschen und anderen Spezies 6,20. Bei Verwendung dieser Methode mit nicht-humanen Isolate, kann es notwendig sein , mit DNA – Oligos zu ergänzen , die komplementär zu den divergierenden rRNA – Sequenzen der spezifischen Wirts 3,21.
Da das Protokoll unvoreingenommen ist, liest virale nur einen kleinen Bruchteil der gesamten Bibliothek Inhalt darstellen. Obwohl rRNA ist die häufigste Art des Wirts RNA und nur ein kleiner Prozentsatz der rRNA liest (0; 1%) werden nach der selektiven Erschöpfung, alle anderen Wirts – RNA (zB mRNA) bleiben nach Erschöpfung und Konto kann für viele Sequenzierung liest aus der Probe gefunden. Daher "Überabtasten" (dh oversequencing) wird einzelne Bibliotheken benötigt , um für Anrufe virale Montage und Variante genügend Deckung zu haben. Für unsere Studien versuchen wir zu Folge ~ 20 Millionen liest pro Probe 2,5 ausreichende Tiefe für die Analyse von viralen genomischen und die damit verbundenen Varianten sowie metagenomic Inhalt zu haben. Für metagenomic und Studien Entdeckung pathogen ist es wichtig, dass kontaminierende Wirts-DNA zu beachten, durch DNase-Verdau entfernt. Daher Viren und andere Pathogene, die DNA-Genome enthalten kann während des Prozesses jedoch RNA Zwischenprodukte können noch sequenzierenden verloren.
Disclosures
The authors have nothing to disclose.
Acknowledgements
This work has been funded in part with Federal funds from the National Institutes of Health, Office of Director, Innovator (No.: DP2OD06514) (PCS) and from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under Contracts (No:HHSN272200900018C, HHSN272200900049C and U19AI110818).
Materials
| 96-Well PCR Plates | VWR | 47743-953 | |
| Strips of Eight Caps | VWR | 47745-512 | |
| Nuclease-free water | Ambion | AM9937 | 50 ml bottle |
| TURBO DNase | Ambion | AM2238 | post RNA extraction step, 2 U/µL, buffer included |
| PCR cycler | any PCR cyclers | ||
| Agencourt RNAClean XP SPRI beads | Beckman Coulter Genomics | A63987 | beads for RNA cleanup |
| Real Time qPCR system | any system | ||
| DynaMag-96 Side Skirted Magnet | Invitrogen | 12027 | |
| 70% Ethanol | prepare fresh | ||
| qRT-PCR primers | IDT DNA | see Table 2 | |
| 5M NaCl | Ambion | AM9760G | |
| 1M Tris-HCl pH 7.4 | Sigma | T2663-1L | |
| 1M Tris-HCl pH 7.5 | Invitrogen | 15567-027 | |
| 1M MgCl2 | Ambion | AM9530G | |
| Linear acrylamide | Ambion | AM9520 | |
| DNA oligos covering entire rRNA region | IDT DNA | see Table 3, order lab-ready at 100 µM | |
| Oligo (dT) | IDT DNA | 40 nt long, desalted | |
| Hybridase Thermostable RNase H | Epicentre | H39100 | |
| RNase-free DNase Kit | Qiagen | 79254 | post selective depletion step |
| SUPERase-In RNase Inhibitor | Ambion | AM2694 | |
| Random Primers | Invitrogen | 48190-011 | mostly hexamers |
| 10 mM dNTP mix | New England Biolabs | N0447L | |
| SuperScript III Reverse Transcriptase | Invitrogen | 18080-093 | with first-strand buffer, DTT |
| Air Incubator | any air incubator cyclers | ||
| NEBNext Second Strand Synthesis (dNTP-free) Reaction Buffer | New England Biolabs | B6117S | 10x |
| E. coli DNA Ligase | New England Biolabs | M0205L | 10 U/μl |
| E. coli DNA Polymerase I | New England Biolabs | M0209L | 10 U/μl |
| E. coli RNase H | New England Biolabs | M0297L | 2 U/μl |
| 0.5M EDTA | Ambion | AM9261 | |
| Agencourt AMPure XP SPRI beads | Beckman Coulter Genomics | A63881 | beads for DNA cleanup |
| Elution Buffer | Qiagen | 10 mM Tris HCl, pH 8.5 | |
| Quant-iT dsDNA HS Assay Kit | Invitrogen | Q32854 | |
| Qubit fluorometer | Invitrogen | Q32857 | |
| Nextera XT DNA Sample Prep Kit | Illumina | FC-131-1096 | |
| Nextera XT DNA Index Kit | Illumina | FC-131-1001 | |
| Tapestation 2200 | Agilent | G2965AA | |
| High Sensitivity D1000 reagents | Agilent | 5067-5585 | |
| High Sensitivity D1000 ScreenTape | Agilent | 5067-5584 | |
| BioAnalyzer 2100 | Agilent | G2939AA | |
| High Sensitivity DNA reagents | Agilent | 5067-4626 | |
| Library Quantification Complete kit (Universal) | Kapa Biosystems | KK4824 | alternative to tapestation, bioanalyzer for library quantification |
References
- Stremlau, M. H., et al. Discovery of novel rhabdoviruses in the blood of healthy individuals from West Africa. PLoS Negl Trop Dis. 9, e0003631 (2015).
- Gire, S. K., et al. Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. Science. 345, 1369-1372 (2014).
- Morlan, J. D., Qu, K., Sinicropi, D. V. Selective depletion of rRNA enables whole transcriptome profiling of archival fixed tissue. PLoS One. 7, e42882 (2012).
- Park, D. J., et al. Ebola Virus Epidemiology, Transmission, and Evolution during Seven Months in Sierra Leone. Cell. 161, 1516-1526 (2015).
- Andersen, K. G., et al. Clinical Sequencing Uncovers Origins and Evolution of Lassa Virus. Cell. 162, 738-750 (2015).
- Matranga, C. B., et al. Enhanced methods for unbiased deep sequencing of Lassa and Ebola RNA viruses from clinical and biological samples. Genome Biol. 15, 519 (2014).
- Tang, F., et al. RNA-Seq analysis to capture the transcriptome landscape of a single cell. Nat Protoc. 5, 516-535 (2010).
- Jiang, L., et al. Synthetic spike-in standards for RNA-seq experiments. Genome Res. 21, 1543-1551 (2011).
- . Kapa Biosystems Available from: https://www.kapabiosystems.com/product-applications/products/next-generation-sequencing-2/library-quantification/ (2015)
- . Illumina Technologies Available from: https://support.illumina.com/content/dam/illumina-support/documents/documentation/system_documentation/miseq/preparing-libraries-for-sequencing-on-miseq-15039740-d.pdf (2015)
- Kircher, M., Sawyer, S., Meyer, M. Double indexing overcomes inaccuracies in multiplex sequencing on the Illumina platform. Nucleic Acids Res. 40, 3 (2012).
- Trombley, A. R., et al. Comprehensive panel of real-time TaqMan polymerase chain reaction assays for detection and absolute quantification of filoviruses, arenaviruses, and New World hantaviruses. Am J Trop Med Hyg. 82, 954-960 (2010).
- Hu, Y., et al. Serial high-resolution analysis of blood virome and host cytokines expression profile of a patient with fatal H7N9 infection by massively parallel RNA sequencing. Clin Microbiol Infect. 21, 713 (2015).
- Simon-Loriere, E., et al. Distinct lineages of Ebola virus in Guinea during the 2014 West African epidemic. Nature. 524, 102-104 (2015).
- Folarin, O. A., Happi, A. N., Happi, C. T. Empowering African genomics for infectious disease control. Genome Biol. 15, 515 (2014).
- Blainey, P. C., Quake, S. R. Digital MDA for enumeration of total nucleic acid contamination. Nucleic Acids Res. 39, 19 (2011).
- Malboeuf, C. M., et al. Complete viral RNA genome sequencing of ultra-low copy samples by sequence-independent amplification. Nucleic Acids Res. 41, 13 (2013).
- Gonzalez, I. L., Sylvester, J. E., Smith, T. F., Stambolian, D., Schmickel, R. D. Ribosomal RNA gene sequences and hominoid phylogeny. Mol Biol Evol. 7, 203-219 (1990).
- Adiconis, X., et al. Comparative analysis of RNA sequencing methods for degraded or low-input samples. Nat Methods. 10, 623-629 (2013).
