To validate the Circle-Seq method, three S. cerevisiae CEN.PK populations of 1 x 1010 cells were screened after cells were grown separately in YPD for ten generations. Chromosomal linear DNA elimination was confirmed by the absence of a qPCR ACT1 signal as described previously20 (data not shown). Purified and enriched eccDNA was sequenced up to 68 million reads (141-nucleotide single-end reads) and mapped to the CEN.PK113-7D reference genome (version 19 June 2012). Recordings of putative eccDNAs from the three samples named C1, C2 and C4 were assigned to genomic regions mapped by contiguous reads longer than 1 kb. Based on 10,000 Monte Carlo simulations, the significance of each region mapped by contiguous reads longer than 1 kb was estimated. From this 79, 159 and 56 regions were annotated as likely eccDNA sequences (p < 0.1, Dataset 1). The number of recorded contiguous reads > 1 kb increased as a function of sequence depth suggesting that even more eccDNA elements would have been recorded if samples had been sequenced further (Figure 2). As expected, the Circle-Seq method extracted numerous reads from a number of known circular DNA elements including the 2µ plasmid, mitochondrial DNA, ribosomal RNA genes on chromosome XII, and the three internal control plasmids pBR322, pUC19 and pUG72 that were spiked into samples just before column purification (Figure 3).
The video shows an example of contiguous reads that mapped to the HXT7_ARS432_HXT6 locus on chromosome IV. Previously, the [HXT6/7circle] was detected by Circle-Seq in ten S288c populations (each with 1 x 1010 cells) and the circular DNA structure was confirmed by inverse PCR analysis20. The [HXT6/7circle] was also recorded in each of the three CEN.PK populations (Figure 4A). Moreover, most of the common eccDNA genes among replicate samples of CEN.PK overlapped eccDNA genes from the S288c datasets (Figure 4B).
To test the specificity of the Circle-Seq protocol for circular DNA purification, two samples, each with 30 µg genomic DNA, were tested. One sample was supplemented with 100 ng plasmid DNA and eccDNA from both samples were purified by the Circle-Seq protocol. After column separation, the DNA yield was 1.27% (380 ng) for the sample without plasmid (GD) and 1.60% (480 ng) for the sample with plasmid (GD+P). The efficiency of exonuclease treatment was tested for linear DNA content after 29 hr and 72 hr using PCR against ACT1. No samples contained amplified ACT1 (data not shown). A fraction of each exonuclease-treated sample was further amplified by the ø29 polymerase and the products of enzymatic reactions were analyzed by propidium iodide staining (Figure 5A-F) and agarose gel electrophoresis (Figure 5G). Samples after exonuclease treatment showed minimal propidium iodine-stain (Figure 5A-B). The ø29–amplified sample with only genomic DNA revealed thread-like structures (Figure 5C) similar to the control sample (Figure 5E). The ø29–amplified sample that had added plasmid revealed foci (Figure 5D) resembling the plasmid control (Figure 5F). The images indicated that ø29 polymerase enriched for circular DNA over linear DNA. Most linear chromosomal DNA was removed from samples after 29 hr exonuclease treatment (Figure 5A-B, G). However, extensive exonuclease treatment for more than 100 hr and using more than 100 units was needed to remove all chromosomal linear DNA, as ø29–amplified samples still showed a background of thread-like structures after 72 hr exonuclease treatment (Figure 5C-D).
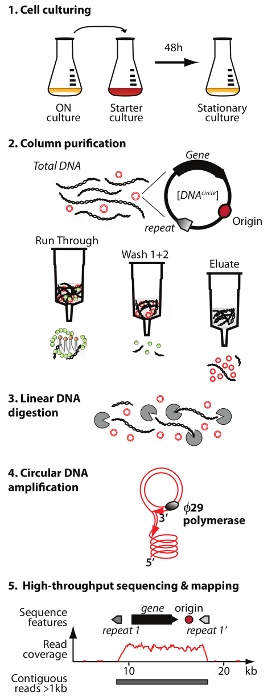
Figure 1. Outline of the Circle-Seq method. The protocol has 5 steps: 1) cell culturing, 2) purification and enrichment of eccDNA by column chromatography, 3) digestion of remaining linear chromosomal DNA in the eluate fraction, 4) amplification of DNA by ø29 DNA polymerase, and 5) sequencing of highly enriched eccDNA and mapping of reads to the S. cerevisiae reference genome. Please click here to view a larger version of this figure.
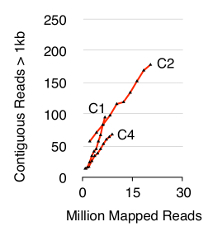
Figure 2. Contiguous reads > 1 kb as function of sequence depth. EccDNA from 1 x 1010 cells increase as a function of sequence depth (in millions of mapped reads). Shown: biological triplicates from haploid CEN.PK S. cerevisiae populations (C1, C2, C4) separated by 1010 cell divisions. Please click here to view a larger version of this figure.
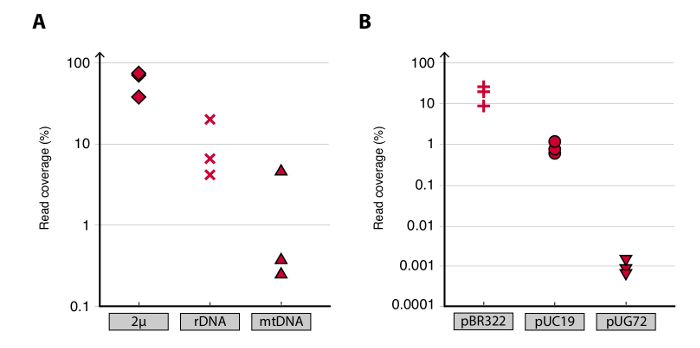
Figure 3. Detection of known circular DNA elements. (A-B) Scatter plots of read coverage (read density) in percent for plasmids in CEN.PK biological replicates C1, C2 and C4. (A) Mapped reads to the endogenous yeast plasmids were: 2µ; [rDNAcircle] (ribosomal RNA genes from chromosome XII); and mtDNA (the mitochondrial DNA). (B) Unique reads mapped to control plasmids. Control plasmids were spiked into samples before column purification. Plasmid ratios per cell were: pBR322 (plus signs) 1:1, pUC19 (circles) 1:50, and pUG72 (triangles) 1:2,500.
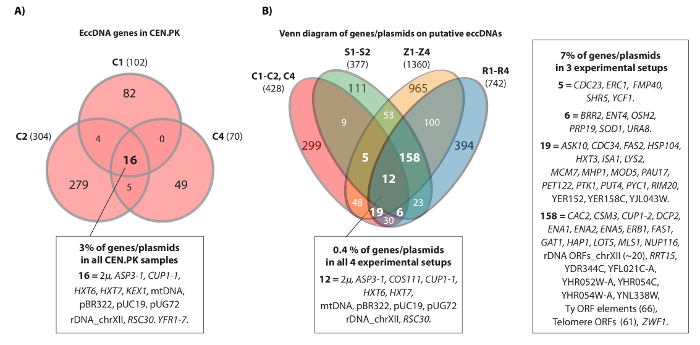
Figure 4. Common eccDNA elements in CEN.PK and S288c. (A) Venn diagram displaying overlap among the 476 genes on 294 eccDNA elements in the three CEN.PK samples (C1, C2, C4). The 16 common overlapping eccDNA genes/plasmids are annotated (all gene names are in Dataset 1). (B) Venn diagram of all recorded genes on putative eccDNAs from the three CEN.PK samples (C1, C2, C4), compared to all recorded genes on putative eccDNAs from 10 S288c samples: S1-S2, R1-R4, Z1-Z4 (see reference20). Shown are 13 biological replicates (S1-S2, R1-R4, Z1-Z4, C1-C3) with genes/plasmids and putative eccDNA regions that overlapped a minimum of 2 strain backgrounds and either 3 or more experimental setups. C samples, CEN.PK; R and Z samples, S288c BY4741; S samples, S288c M3750. Please click here to view a larger version of this figure.
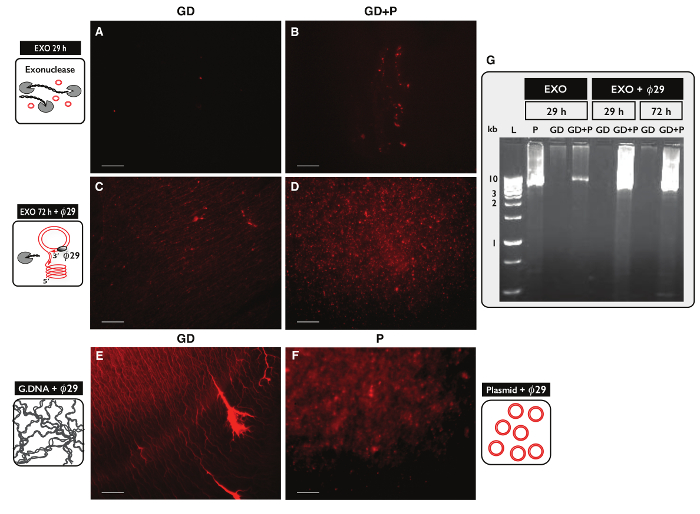
Figure 5. Visualization of DNA samples after exonuclease and ø29 treatment. (A-F) Propidium iodide staining of DNA. Scale bar, 10 µm. (A, C and E) Samples with genomic DNA (GD); (B and D) samples with GD plus plasmid (GD+P). (A-B) After 29 hr exonuclease treatment (EXO 29 h); (C-D) after 72 hr exonuclease treatment followed by ø29 polymerase amplification (EXO 72 h + ø29). (E) Genomic DNA control after e: ø29 polymerase amplification; (F) plasmid control (5.5 kb) after 29 polymerase amplification; (Gø) agarose gel-eletrophoresis. From left: L, 1 kb markers; P, plasmid control (5.5 kb) after EXO 29 hr; GD, after EXO 29 hr (sample as in A); GD+P, after EXO 29 hr (sample as B); GD and GD+P, after EXO 29 hr + ø29; GD and GD+P, after EXO 72 hr + ø29 (sample as in C-D). See Table S1 for extra details. Please click here to view a larger version of this figure.
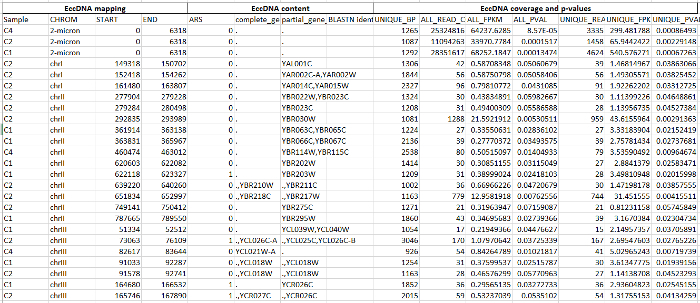
Dataset 1. Potential DNA circularization regions in CEN.PK. Please click here to download this file.
Shown are sequence data and analyses for 348 regions. Columns are A-D, eccDNA mapping. A (first column from left), sample from which putative eccDNA was identified; B, chromosome; C-D, start and end coordinates of putative eccDNAs. E-H, eccDNA content. E, autonomously replicating sequence (ARS) in the region; F, complete gene in the region; G, part of gene included in the region; H, BLASTN-identified gene. I-O, EccDNA coverage and p-values. I, longest region with a uniquely annotated sequence in bp; J, number of all mapped reads; K, coverage of all mapped reads by fragments per kb from a million mapped reads (FPKM); L, p-value for putative eccDNA compared to occurrence by chance from Monte Carlo simulations; M, number of uniquely mapped reads; N-O; as K and L using only uniquely mapped reads (UFPKM). Parameters for mapping of reads and Monte Carlo simulations were as described20.
