El proceso de transferencia de protones en porficéos tiene aplicaciones potenciales en el desarrollo de interruptores moleculares, transistores y dispositivos de almacenamiento de información1,2. En particular, la tautomerización en porficéos a través del proceso de transferenciade doble protones ha atraído un amplio interés en los campos de la espectroscopia y la fotofísica 2. Los átomos de hidrógeno interno de porficeno pueden migrar de un isómero trans al otro isómero trans equivalente a través del proceso de transferencia de doble protones como se muestra en la Figura 1. Se han propuesto dos mecanismos para el proceso de transferencia de doble protones: el mecanismo concertado y el mecanismo escalonado3,4. En el proceso concertado de transferencia de doble protones, ambos átomos de protones se mueven al estado de transición sincrónicamente de una manera simétrica, mientras que un protón completa la transferencia antes que el otro protón en un proceso escalonado. Dos átomos de hidrógeno pueden transferirse simultáneamente o escalonadamente dependiendo de la fuerza de correlación entre dos átomos de hidrógeno5.
La sustitución isotópica se ha utilizado para detectar las propiedades estructurales de las moléculas y velocidad constantes de la cinética de reacción6. La sustitución de deuterio único en el hidrógeno interno del porficeno conduce a una forma asimétrica de la molécula. El enlace de hidrógeno puede expandirse o contraerse debido a la diferencia de masa entre los átomos de hidrógeno y deuterio. La sustitución isotópica introduce una perturbación en el andamio del porficeno. Se plantea la cuestión de si la estructura asimétrica afectaría el proceso de transferencia de protones. Limbach y compañeros de trabajo informaron que la sustitución del hidrógeno por deuterio comprimirá tanto los enlaces de hidrógeno, como el acoplamiento cooperativo de dos enlaces de hidrógeno en porficeno puede favorecer el mecanismo concertado7,mientras que Yoshikawa declaró que el deuteración haría que el mecanismo escalonado contribuyera más que el mecanismo concertado8. Se han desarrollado técnicas experimentales, como la espectroscopia de fuerza, paracapturar detalles de tautomerización en un solo porficeno 9. Sin embargo, sigue siendo difícil determinar los detalles atómicos de la transferencia de protones experimentalmente debido a su naturaleza transitoria.
Los cálculos teóricos y las simulaciones pueden actuar como herramientas complementarias para esclarecer los mecanismos de reacción de la transferencia de protones. Entre los diferentes métodos teóricos, las simulaciones de dinámica molecular (MD) pueden monitorear los movimientos dinámicos de cada átomo, y se ha utilizado ampliamente para revelar mecanismos complejos en reacciones químicas y enzimáticas. Sin embargo, las simulaciones MD regulares tienden a sufrir problemas de muestreo insuficientes, especialmente cuando existe una barrera de energía alta en el proceso de interés. Por lo tanto, se han desarrollado métodos de muestreo mejorados, que incluyen el muestreo de ruta de transición10,11, muestreo paraguas (EE.UU.)12,13, y muestreo de templado integrado (ITS)14, 15. La combinación de diferentes métodos de muestreo mejorados puede aumentar aún más la eficiencia de muestreo16,17,18. Para aprovechar los algoritmos de muestreo mejorados en la simulación de reacciones químicas, hemos implementado el método de muestreo de templado integrado selectivo (SITS) con potencial mecánico cuántico y mecánico molecular (QM/MM) recientemente19. El método SITS-QM/MM propuesto combina las ventajas de ambos métodos: el método SITS acelera el muestreo y puede explorar todos los canales de reacción posibles sin conocimiento previo del mecanismo de reacción, y QM/MM proporciona una descripción más precisa de el proceso de formación de unión y ruptura de unión, que no puede ser simulado por los métodos MM únicamente. El enfoque SITS-QM/MM implementado ha descubierto con éxito la transferencia concertada de doble protones, mecanismo de transferencia de doble protones no correlacionado y correlacionado en diferentes sistemas, sin predefinir las coordenadas de reacción19. En el caso del porficeno, se ha notificado el carácter de transferencia de protones escalonapero pero correlacionado19. El método híbrido SITS-QM/MM se utilizó para investigar el efecto isotópico en el porficeno en nuestro estudio, y a continuación se muestran las descripciones detalladas del algoritmo y el protocolo de nuestro método.
Hemos implementado el método SITS con potencial sin QM/MM híbrido. Se definió el potencial efectivo de SITS para incluir la energía potencial a diferentes temperaturas con los factores de ponderación nk para cubrir rangos de temperatura más amplios,
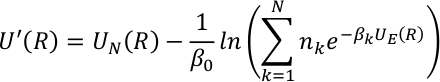
donde, N es el número de términos canónicos, la temperatura inversa es la temperatura inversa, y nk es el factor de ponderación correspondiente para cada componente canónico. UE (R) y UN(R) representan los términos mejorados y no mejorados en SITS y se definen como,
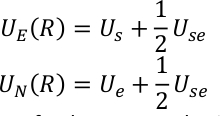
U s, Use y Ue son la energía potencial del subsistema, la interacción entre el subsistema y el medio ambiente, y la energía potencial del medio ambiente. El potencial QM/MM se expresa como una suma híbrida de tres componentes,
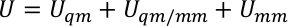
donde Uqm, Uqm/mmy Umm son el término de energía interna del subsistema QM, la energía de interacción entre las regiones QM y MM y la energía de interacción dentro del subsistema MM, respectivamente. El término Uqm/mm se puede dividir en tres componentes, que incluyen los términos electrostáticos, van der Waals y energía de interacción covalente entre los átomos QM y MM,
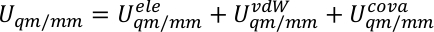
Asignamos, 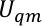
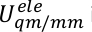 y en un término de Us en SITS,
y en un término de Us en SITS,
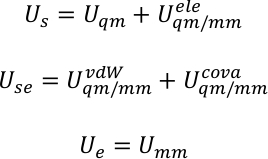
El potencial total del sistema se descompuso entonces en la energía del subsistema U s, la energía de interacción entre el subsistema y el medio ambiente Use, y la energía del medio ambiente Ue. Por ejemplo, en el sistema de la obra actual, el subsistema es la porficencia, y el medio ambiente el agua.
El perfil PMF a lo largo de una variable colectiva (R) se deriva como,
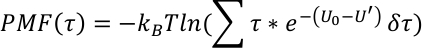
Las coordenadas de reacción generalmente utilizadas para cada transferencia de hidrógeno de N1–H1– N2 son q1 o (r1ar2)/2 y q2 a r1 + r2, donde se encuentran1 es la distancia de N1-H1 y r2 es la distancia de H1-N2.
El método se ha implementado en el paquete de simulación QM/MM MD QM4D20. El código fuente completo y la documentación se pueden encontrar aquí: http://www.qm4d.info/.
Generalmente, las simulaciones SITS-QM/MM MD implican cuatro pasos: pre-equilibrio (pre-sits); optimización nk (opt-sits); simulación de producción y análisis de datos.
