La versatilidad de la caja de herramientas QSAR de la OCDE como software analítico para la ecotoxicología se muestra aquí con un interés específico en los efectos adversos de los productos químicos disruptores endocrinos en los vertebrados acuáticos. Además, se demostró un protocolo simple y estándar para predecir la toxicidad aguda (96-h LC50) de 74 EDs representativos (Tabla1) para las especies de peces. Esto se logró aplicando módulos de creación de categorías, llenado de brechas de datos y generación de perfiles de ER integrados en el cuadro de herramientas QSAR (Figura1, Figura 2).
La correlación lineal entre el registro10LC50 y el registro10KOW con una pendiente negativa (como se muestra en la figura suplementaria S1)ha sido conocida durante mucho tiempo como una relación cuantitativa estándar en los análisis QSAR25,donde mayor toxicidad se muestra cuanto más hidrófobo es un producto químico dado. Como se puede ver en un simple cálculo, la relación matemática general que incluye la Ecuación S1 y la Ecuación S2 (InformaciónSuplementaria)es una expresión convertida de la siguiente función de potencia26:
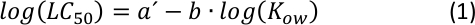

A partir de la gráfica de(Ecuación 2), caracterizar un rango intermedio de KOW26 puede ser posible ajustando los parámetros a y b, donde una cierta variación en hidrofobicidad (o hidrofilicidad) no cambia significativamente la punto final de toxicidad aguda.
Los análisis comparativos entre las predicciones computacionales y las observaciones experimentales sobre la LC50, como se muestra en la Figura 3 y la Figura 4,se han notificado típicamente en estudios de QSAR para diversos tóxicos acuáticos, incluyendo tensioactivos técnicos no iónicos27, fungicidas triazoles28y metabolitos plaguicidas21. Este tipo de validación retrospectiva proporciona información sobre hasta qué punto una determinada herramienta QSAR puede llegar en términos de rendimiento comparativo a los resultados experimentales. En este estudio de toxicidad aguda en peces, se demostró que QSAR Toolbox proporciona predicciones protectoras para más del 90% de los ED probados en todos los peces y en una sola especie, Pimephales promelas.
Se requiere una mayor identificación de los tres productos químicos atípicos en la Figura 3 y la Figura 4, que mostraron una LC50 pronosticada más alta en promedio y como mínimo, respectivamente. En primer lugar, el 3′,5,7-trihydroxy-4′,6-dimethoxyisoflavone es un tipo de flavonoide (más específicamente, una isoflavona), que se considera generalmente seguro y se utiliza en productos farmacéuticos herbarios; sin embargo, todavía tiene preocupaciones relacionadas con el estrógeno29 y puede causar toxicidad aguda probablemente a través de desacoplamiento de fosforilación oxidativa30. A continuación, el 1,4-benzenediol, llamado hidroquinona, es un compuesto fenólico que puede desencadenar una respuesta inmune no específica y citotóxica en los peces31. Finalmente, el 4-hexylphenol se ha sabido para exhibir suficiente actividad estrogénica positiva para ser clasificado como un ED32. Se ha estudiado bien que la razón principal de la toxicidad aguda de la hidroquinona es el ciclo de reducción-oxidación (redox). La hidroquinona se oxida a benzoquinona y se reduce de nuevo a semiquinona o hidroquinona repetidamente, con cofactores que agotan y generando especies reactivas de oxígeno33. Los otros dos productos químicos pueden requerir investigaciones más profundas para revelar sus mecanismos de acción en ecotoxicidad aguda utilizando enfoques de acoplamiento molecular como el utilizado por Panche et al.34, que no pueden ser cubiertos por la caja de herramientas QSAR.
Los ED interfieren con el sistema endocrino principalmente a través de interacciones fisicoquímicas con receptores de esteroides como los receptores de estrógeno y andrógenos, que son de considerable interés en los estudios de modelado QSAR35. Teniendo en cuenta esto, QSAR Toolbox es robusto en términos de clasificación fácil y rápida de afinidades de unión er para un conjunto de productos químicos basados únicamente en los descriptores 2D de las estructuras moleculares. Cuando este sistema de generador es de perfiles de ER se aplicó a nuestra lista de EDs, no se encontró una correlación clara entre la afinidad de unión de ER y la hidrofobicidad (FiguraSuplementaria S2). Este resultado puede explicarse por el hecho de que la formación de un complejo esteroide-receptor no es una consecuencia directa de una contribución de unión hidrófoba, sino que debe ir acompañada de un cambio de conformación en la estructura del receptor del sitio activo36. La unión del receptor también puede deberse a la unión al hidrógeno y al apilamiento de la s.
Además, la posición de cada grupo químico en la molécula puede afectar a la unión del receptor, incluso si la hidrofobicidad y el número de receptores-bonos de hidrógeno siguen siendo los mismos. En segundo lugar, el generador de perfiles de ER produjo tendencias contrarias entre los niveles medios de LC50 previstos y experimentales con una creciente afinidad de unión de ER (Figura5). Esto puede deberse a que la letalidad de los padres en una prueba de toxicidad aguda no se debe a la unión a Urgencias, sino más bien a la narcosis en la mayoría de los casos, o al ciclo de redox en el caso de la hidroquinona. Por ejemplo, se requiere un análisis más extenso, incluida la toxicidad crónica, para que un conjunto más grande de EDs defina las limitaciones predictivas de la versión actual de QSAR Toolbox.
Esta investigación preliminar también puede tener implicaciones para la salud pública porque los esteroides (andrógenos, estrógenos, progestinas, y corticoides) y sus receptores exhiben estructuras macromoleculares similares o incluso idénticas a través de vertebrados5. Estos tipos de sistemas de señalización endocrino análogos pueden funcionarutilizando un mecanismo común en eventos clave de los DATOS Electrónicos 5. Sin embargo, se requieren metodologías adicionales y complementarias para iluminar este vasto y complejo aspecto [por ejemplo, mediante la realización de modelos computacionales de absorción, distribución, metabolismo y excreción (ADME) y/o resultados adversos vía (AOP)]38. Además, debido a que la mayoría de las preocupaciones científicas y públicas planteadas sobre los efectos adversos de los DEE están relacionadas con sus toxicidades crónicas, mejorar las bases de datos y algoritmos en la caja de herramientas QSAR y producir ecotoxicología fiable a largo plazo las predicciones de los DE Son necesarias.
Este artículo demuestra la aplicación de QSAR Toolbox para comparar los valores ecotoxicológicos de LC50 para peces con los valores de registro10Kow de EDs. A lo largo del protocolo, resulta en relaciones débiles entre los dos parámetros, ya que ha sido revelado por estudios anteriores (por ejemplo, Kim et al.39) que el registro10Kow no es un buen predictor directo de LC50acuático. A pesar de esta limitación, este protocolo proporciona una revisión general o “viñeta” para describir cómo utilizar el panel para un propósito determinado, ya que es una aplicación válida para utilizar qSAR Toolbox para investigar correlaciones entre LC50 (o enlace ER afinidad) y el registro de10Kow, o como una herramienta para la detección rápida de ecotoxicidad aguda. No obstante, cabe señalar que (1) iluminar el vínculo entre la unión del receptor de estrógeno y la toxicidad crónica, en lugar de la toxicidad aguda (letalidad), es más relevante para que se puedan encontrar correlaciones más claras, y (2) el receptor de andrógenos, con la de estrógeno, también juega un papel crítico en la toxicidad reproductiva. Por lo tanto, es necesario que la versión futura del cuadro de herramientas QSAR mejore las funciones de predicción a la luz de esos dos puntos.
