Validation of optimum cell growth on chip
A major goal of the experiment platform is to reproduce key components and interactions in a complex tumor microenvironment in a comprehensive manner, yet simple enough to provide quantitative, reliable and reproducible data. This goal can only be achieved if we have full control of the physical and biochemical environmental factors. We must either exclude the undesired factors or figure out a way to incorporate the uncontrollable factors into the quantitative model to correctly interpret the population behaviors.
The first test is to ensure optimum cell growth on the device. The cell proliferation rate on the device needs to be identical or comparable to their growth profile in conventional cell culture. As a proof of principle, human prostate cancer cell line PC3 cells were cultured in the PDMS chip without the presence of external stress. These cells were fluorescently-labeled in the cytoplasm so we could observe and quantify the population of cells using inverted fluorescent microscopy. Cells confluence, defined as the area that covered by cells, was measured at each microhabitat at different time frames given a selected intensity threshold on the fluorescent images using Fiji15. The growth curves of cells at each chosen position are plotted in Figure 4. The chosen positions are scattered across the chip, from the region near the apex of media inflow to the apex close to media outlets.
The growth curves in Figure 4B are well-aligned, suggesting the identicality of the cell proliferation profile as a function of position across the chip. The initial slope of the curves reveals the proliferation rate. The doubling time for cells in the device is around 24 hours as shown in Figure 4B from time 0 to 24 hours, which is the same as the doubling time in conventional cultureware13. Therefore, we conclude that without the presence of an external source of stress, the physical and biochemical factors are uniformly distributed, leading to the homogeneous and optimized cell growth from time 50 to 100 hours.
We further considered the ability to quantify cell dynamics between subpopulations in a microenvironment. GFP-expressing PC3-EMT and the mCherry-expressing PC3-EPI cell lines were co-cultured in the EA as shown in Figure 5. Cell confluence, defined as the percentage of area covered by the cells, is measured as the indication of the size of population. Starting from 40% confluence, the co-culture grew fully confluent within 3 days. Interestingly, we may observe the progression of each phenotype in the microenvironment. While the growth curve of total confluence is in the form of logistic growth model, the population of PC3-EMT continued to expand and PC3-EPI was suppressed in the asymptotic phase.
Demonstration of cell motility assay in a mixed population
Cell motility is an important phenotypic behavior. The motility distribution of a population can provide information about the physical interaction of cells, the mechanical properties of local microenvironment, and can sometimes be analyzed as an indication of epigenetic variation of cells. Given that our improved cell culture platform allows nearly continuous imaging, the advantage of having fine temporal resolution maximizes the potential of single cell tracking in a heterogeneous population.
We use the plugin TrackMate16 in Fiji (http://fiji.sc) with customized macro to implement and automate the tracking process. TrackMate allows the user to implement and customize a tracking algorithm using a scripting language from the Fiji Script Editor. The tracking process is divided in a series of steps: the segmentation, filtering, and particle-linking processes.
As shown in Figure 6B, we performed a "wound healing assay" as a demonstration to study cell collective migration and cell-cell interaction. PC3-EPI and PC3-EMT cells were densely seeded in the center of the device and were allowed to freely migrate toward empty region. The normalized histogram of the speed of both phenotypes is plotted in Figure 6C. The velocity of PC3-EMT was significantly higher than PC3-EPI by a factor of 1.8x, which is consistent with the fact that the mesenchymal phenotype serves as a component of cutaneous wound healing with exceptional ability to migrate as opposed to the comparably stationary epithelial phenotype.
Further, in order to observe the progression of cell motility in a stress-free environment, we performed a long-term co-culture of PC3-EPI and PC3-EMT with uniform initial seeding density and tracked the distribution of cell velocity varying with time. As shown in Figure 7A,B, after 6 days of culture, as cells reached higher confluence, the distributions of cell speed for both phenotypes leaned toward the left tails.
We may compare the ratio of number of PC3-EPI to PC3-EMT in each class interval. As shown in Figure 7C, regardless of the days of culture or density of cells, PC3-EMT dominated the high-speed region while PC3-EPI occupied the low speed zone. Although the overall velocity decreased significantly for both phenotypes, PC3-EMT remained motile and was affected less by the crowdedness of the microenvironment as shown in Figure 7D. The average speed of PC3-EPI dropped around 40% while that of PC3-EMT only dropped 14%.
There are several factors that can affect the distribution of cell velocity and the average speed, such as the crowdedness of the population. Continuous cell tracking and monitoring over all subpopulation is essential to understand the progression of phenotypic behavior. For example, if we would like to quantify the level of the mesenchymal status of an unlabeled subpopulation, it would be more informative to acquire not only the distribution of the physical quantity (e.g., velocity) of the interested subpopulation as a function of time, but also that of all the rest of the coexisting subpopulation of cells.
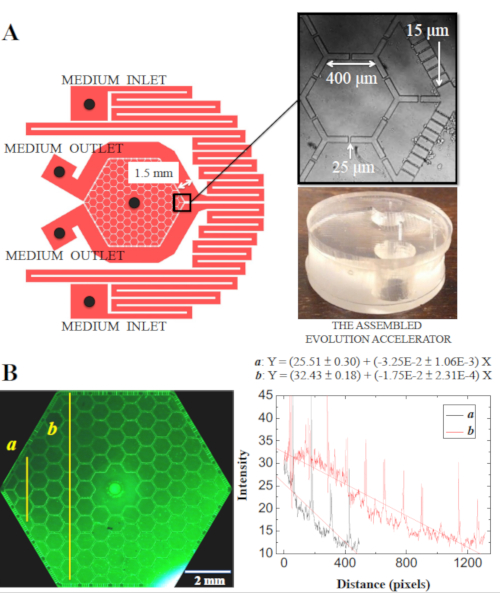
Figure 1: The syringe-pump-driven microfluidic EA chip. (A) The microfluidic device has two separate medium inlets, two outlets, and one central port for cell seeding or extraction of excessive medium. Note that between the medium inlets and the cell culture arrays, a pair of serpentine channels that allow the medium to equilibrate with the atmosphere passed below the gas-permeable membrane. (B) The generation of gradient. The syringes pump the medium with ~ 0.1 mM of fluorescein and no fluorescein respectively into the chip through the 2 medium inlets on the right, extracted out from the chip at the 2 medium outlets on the left. The concentration of fluorescein is proportional to the intensity of fluorescence signal, and is profiled near the medium inlets/outlets a and near the center b. Figure reproduced with permission from Lin et al. 201712. Please click here to view a larger version of this figure.

Figure 2: The components of the customized 3 well sample plate. (a) the main body of 3 well plate; (b) a pair of gas channels that allow conditioned atmosphere to be pumped in and vented out through the septa at the entrance of the gas channels; (c) an O-ring designed to seal the space between the well plate and the gas-permeable culture dish; (d) the 35 mm diameter gas-permeable culture dish; (e) the dish holder; (f) the PDMS device; (g) the PDMS chip holders; (h) the double layer 35 mm glass windows designed to maintain thermal isolation and prevent water condensation; (i) glass window holder; (j) heating pads; (k) temperature sensor; (l) the Adhesive sealer that keeps the chip from drying out. Figure reproduced with permission from Lin et al. 201712. Please click here to view a larger version of this figure.
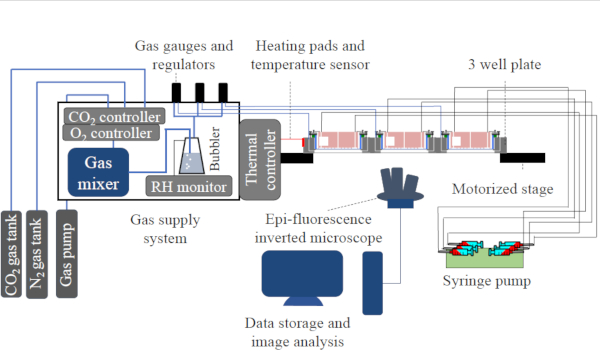
Figure 3: The schematic figure of the experimental setup. The setup of the experiment, including the gas supply system, gas channels connection, the medium supply connection and the imaging system. Figure reproduced with permission from Lin et al. 201712. Please click here to view a larger version of this figure.
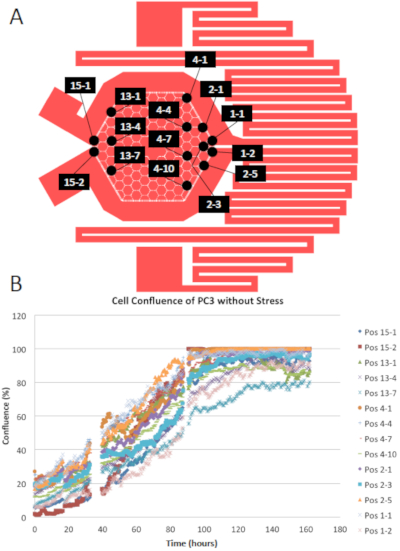
Figure 4: Growth curves of PC3 in the EA without stress. PC3 were cultured in EA without the presence of external stress. (A) Pattern of EA with position labels. (B) Growth curves of PC3 at the chosen positions in terms of cell confluence. Please click here to view a larger version of this figure.
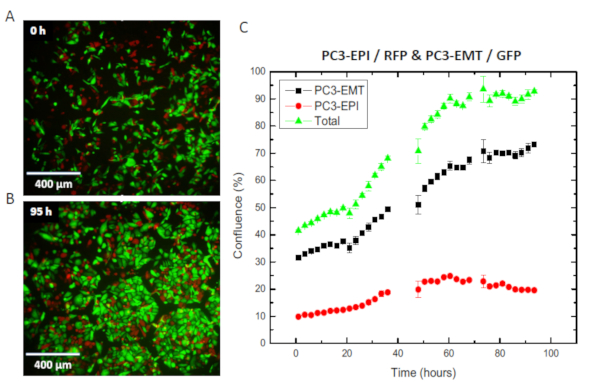
Figure 5: The co-culture of PC3-EPI and PC3-EMT in the EA without stress. PC3-EPI (red) and PC3-EMT (green) were co-cultured in the EA without the presence of external stress. (A) Overlay of two fluorescent channels at t = 0 h. (B) Fluorescent image taken at t = 95 h. (C) Growth curves in terms of cell confluence. Cell confluence of each cell type was measured on the entire chip. Each data point represents the average cell confluence at 3 consecutive time points taken at 15-min intervals. The error bars represent the standard deviation of cell confluence at the 3 consecutive time points. Please click here to view a larger version of this figure.
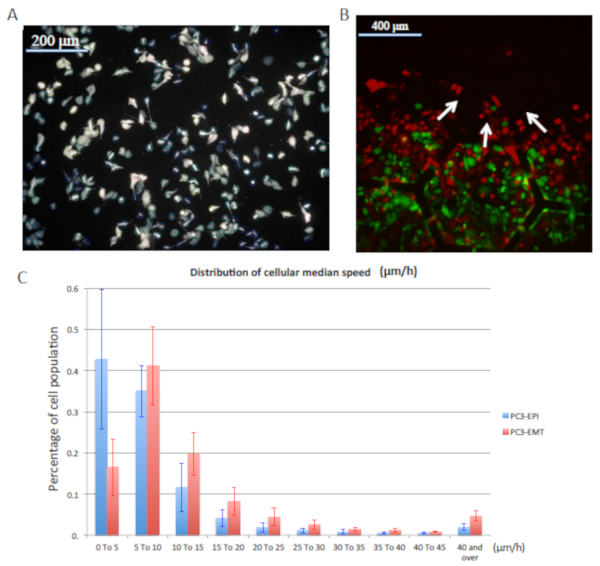
Figure 6: Cell motility assay in a mixed population. The demonstration of single cell tracking performance in a mixed population. (A) A demonstration of how cells are detected by Laplacian of Gaussian (LoG) segmentation. The circles indicate the location of cells detected by the LoG algorithm. The detected cells are linked from frame to frame based on the linear assignment problem to quantify cell motility. (B) The "wound healing assay" of PC3-EPI and PC3-EMT co-culture experiment. Cells were seeded locally up to 100% confluence with a clear boundary at the center. After the installation of the EA chip, cells were observed to migrate through the microhabitat array as indicated by the white arrows. (C) Normalized histogram of cell motility. PC3-EMT is faster than PC3-EPI by a factor of 1.8x. The error bars represent the standard deviation of 5 experiment samples. Please click here to view a larger version of this figure.
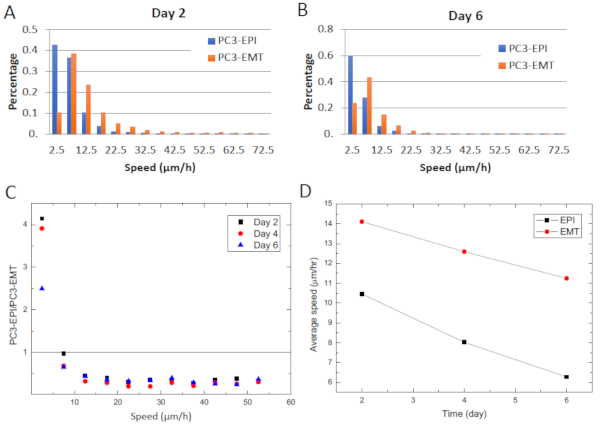
Figure 7: The motility assay of a long-term co-culture of PC3-EPI and PC3-EMT. A long-term co-culture of PC3-EPI and PC3-EMT with uniform initial seeding density. The distribution of cell velocity varies with time. (A) Normalized distribution of cell speed on day 2. (B) The normalized distribution of cell speed on day 6. (C) Ratio of number of PC3-EPI to PC3-EMT in each class interval. (D) Average speed of PC3-EPI and PC3-EMT as a function of time. Please click here to view a larger version of this figure.
