在过去的二十年里,由聚合有机分子制成的固体和薄膜在光电子器件中找到了多种应用。基于此类材料的设备具有许多吸引人的性能,包括重量轻、灵活性强、功耗低,以及使用喷墨打印进行廉价生产的潜力。基于有机发光二极管(OLED)的显示器正在取代液晶显示器,作为手机、笔记本电脑、电视机和其他电子设备11、2、3、42,3,4的先进设备。OLED在照明应用方面的重要性预计在未来4年会增加。有机光伏器件的性能稳步提高,最近单结有机太阳能电池的功率转换效率超过16%。有机材料还有可能破坏其他技术,如光纤通信,其使用使光电调制器的带宽高达15 THz及以上66,7。7
优化光电子应用的固态分子材料的一个主要挑战是,其特性通常在很大程度上取决于材料的纳米级结构。生产过程允许使用受控生长技术(如化学气相沉积、将光学活性分子分泌到另一材料(即聚合物基质9,9、10)、热退火11、12,12等,在一定程度上定义材料的纳米结构。然而,纳米级紊乱是大多数分子材料固有的,通常不能完全消除。因此,了解无序如何影响材料的特性,并找到方法,以达到最佳性能,对于有机光电子材料的合理设计至关重要。
分子材料紊乱的程度通常太大,不能将其视为周期性晶体结构的扰动,其电子结构可以通过波段理论来描述。另一方面,在模拟中必须包括以重现散装材料或薄膜特性的分子数量太大,无法使用密度函数理论(DFT)13、14和时间相关密度函数理论(TD-DFT)15、16等第一原理量子化学方法。13,14 15,16在光电子中的应用有机分子通常具有相对较大的+偶联系统;许多人也有捐助者和接受者团体。捕捉这些分子中正确的电荷转移行为对于计算其光电子特性至关重要,但只能使用TD-DFT17、18、19、20,19,中的远程校正混合功能来完成。17,20使用这种功能的计算与系统的大小线性缩放,目前,它们只可用于对单个有机分子或小分子聚合的光电特性进行建模,这些特性可以使用不超过±104原子基础函数进行描述。一种可以描述由大量色光荷组成的无序材料的模拟方法对于对这些系统的建模非常有用。
分子材料中分子间相互作用的大小通常与构成材料的各个分子之间的能量参数(如能量或激发能量)的变化顺序相当或小于。在这种情况下,多尺度建模是了解和优化大型无序分子系统的光电子特性的最有希望的方法21、22、23。,22,23这种方法使用第一原则量子化学方法(通常是DFT和TD-DFT)来精确计算构成材料的各个分子的特性。然后,使用为单个分子计算的参数构造足够大以表示大分子材料的材料样本的 Hamiltonian(也许,通过使用周期性边界条件)。然后,这个汉密尔顿人可以用来计算大分子聚合、薄膜或大分子材料的光电参数。
Exciton模型是多尺度模型的一个子类,其中分子材料的兴奋状态以兴奋度为基础表示:由库仑吸引力24、25,25绑定的电子孔对。为了模拟许多兴奋状态过程,只包括Frenkel兴奋26,其中电子和孔在同一分子上局部化就足够了。电荷转移冲锋,其中电子和孔是局部在不同的分子上,可能需要包括在某些情况下(例如,建模电荷分离在供体受体系统)27,,28。虽然exciton模型是多尺度模型,只能对单个分子进行第一原理计算,但它们仍然包含分子间相互作用。他们可以解释的两种主要相互作用类型是:(a) 分子之间的兴奋耦合,这些分子具有兴奋剂在分子之间去局部或转移的能力,以及 (b) 通过周围分子上的电荷分布对分子上电子密度的静电极化。我们之前已经表明,这两个因素对于模拟分子聚合的光和光电特性都很重要,例如光学吸收光谱29和第一个超极化30。
在本文中,我们提出了一种用于计算大分子聚集体和散装分子材料的光学光谱和其他光电子特性的异化外物质模型的协议。外行汉密尔顿被认为是一个紧密绑定汉密尔顿24,25,24,
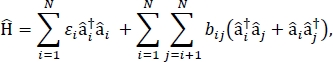
其中#i是物质中ith分子的i激发能量,bij是ith和jthth分子之间的兴奋耦合,[i]†和]我分别是物质中ithth分子的激发状态的创造和毁灭运算符。使用TD-DFT计算在构成材料的单个分子上进行,发现外向汉密尔顿参数。在这些TD-DFT计算中,材料中所有其他分子的电荷分布由静电嵌入原子点电荷来表示,以考虑分子电子密度的静电极化。单个分子的激发能量#i,直接从TD-DFT计算输出中取走。分子之间的外向耦合b bij使用过渡密度立方体方法31计算,具有从高斯32TDFT计算输出和使用多功能波函数分析仪33进行后处理的相互作用分子的接地到兴奋状态过渡密度。为了模拟散装分子固体的特性,周期性边界条件可应用于汉密尔顿。
当前协议要求用户有权访问高斯32和Multiwfn33程序。该协议已经使用高斯16、版本 B1 和Multiwfn版本 3.3.8 进行了测试,但也适用于这些程序的其他最新版本。此外,该协议使用自定义C++实用程序和一些自定义 python 2.7 和 Bash 脚本,源代码在 https://github.com/kocherzhenko/ExcitonicHamiltonian 时在 GNU 通用公共许可证(版本 3)下提供。计算旨在从 Unix/Linux 系列运行操作系统的计算机上执行。
